L’atresia esofagea (AE) è un quadro malformativo caratterizzato dalla mancata formazione del segmento intermedio dell’esofago.
Si distinguono un moncone prossimale, o cervicale, che termina nella maggioranza dei casi (98%) a fondo cieco, ed un moncone distale, in continuità con lo stomaco; quest’ultimo comunica, quasi sempre (86%), con la trachea formando quindi una fistola tracheo-esofagea (FTE).
Epidemiologia
È una malformazione estremamente rara, non prevenibile, che colpisce tra 1:3.000 e 1:10.000 nati vivi. E’ leggermente più frequente nel sesso maschile. È possibile la diagnosi prenatale ma non è facile (solo tramite elementi indiretti come polidramnios e diminuzione del contenuto gastrico). La frequenza di atresia dell’esofago è stata dimostrata essere maggiore nei casi di parti gemellari, ma non c’è nessuna prova genetica.
Qualora non sia stato posto il sospetto diagnostico prenatale, l’età d’esordio clinico della patologia è immediatamente dopo la nascita. Qualora sia presente solo FTE senza AE, l’esordio può essere subdolo e la diagnosi può essere ritardata, anche di mesi.
Embriologia ed eziopatogenesi
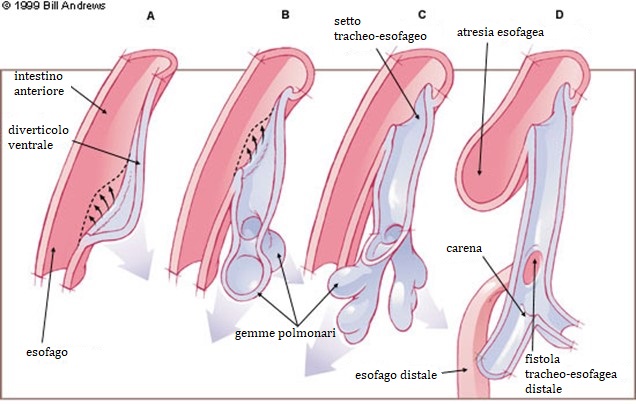
Vie aeree ed esofago derivano da uno stesso progenitore, il tubo intestinale superiore (intestino cefalico primitivo), già presente a 20 giorni dal concepimento. Dalla 4a alla 8a settimana, da tale tubo origina una tasca diverticolare dorsale mediana che dà luogo ad un solco longitudinale bilaterale che rappresenterà la trachea. Avviene così l’allungamento delle due strutture (il tubo laringotracheale posto ventralmente e l’esofago posto dorsalmente) e la separazione di queste tramite un setto tracheoesofageo.
Quello che accade, per cause ancora non del tutto chiare, probabilmente con un’eziologia multifattoriale, è una malformazione per incompletezza di tale processo tra la 3a e 5a settimana di gestazione.
L’atresia esofagea risulta se il setto tracheoesofageo è deviato posteriormente, causando un’incompleta separazione dell’esofago dal tubo laringotracheale (probabilmente causata da eventi vascolari, anomala pressione da parte degli organi circostanti o per difetti di funzione cellulare mesenchimale) e dando origine ad una concomitante fistola tracheoesofagea. L’atresia esofagea senza fistola viene attribuita ad un fallimento della ricanalizzazione dell’esofago durante l’8a settimana di sviluppo embrionale.

Classificazioni
Classificazione di Gross
La classificazione più utilizzata è quella di Vogt, poi completata da Ladd e Gross, che, prendendo in considerazione non solo l’aspetto anatomico della malformazione esofagea, ma anche la presenza o meno di connessioni (fistole) con l’albero tracheo-bronchiale, descrive 5 tipi anatomici:
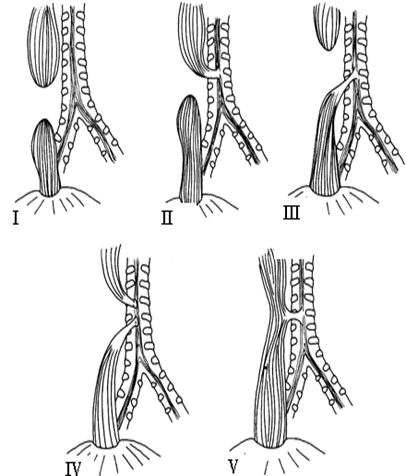 Tipo I (3-5%): AE senza FTE.
Tipo I (3-5%): AE senza FTE.
Totale deconnessione dei due monconi esofagei (long gap) e nessuna comunicazione con l’albero bronchiale.- Tipo II (1-2%): AE con FTE prossimale.
La fistola è generalmente breve e ristretta; origina dalla parete anteriore del moncone esofageo superiore e termina direttamente in trachea. - Tipo III (85-87%): AE con FTE distale.
Si tratta della forma anatomica di più frequente riscontro. La tasca esofagea superiore termina a fondo cieco. L’estremità distale del moncone esofageo comunica con l’albero respiratorio tramite un tragitto fistoloso che si apre a circa 1,5 cm dalla carena stessa o nel bronco principale destro. Esistono 2 varianti:- III A) minore distanza dei 2 capi esofagei, in cui è possibile un’anastomosi primaria;
- III B) maggiore distanza dei 2 capi esofagei, con anastomosi difficoltosa.
- Tipo IV (3-5%): AE con FTE prossimale e distale.
Un tramite fistoloso tra esofago e trachea è presente sia sul moncone prossimale che su quello distale. - Tipo V (3-6%): FTE senza AE.
Si tratta di una forma caratterizzata da una normale anatomia endoluminale dell’esofago ma con la presenza di una fistola tracheo-esofagea (ad N o H). Si presenta tardivamente con sintomi solo respiratori.
Classificazione di Spitz
La classificazione di Spitz è una classificazione prognostica basata sul peso alla nascita e la presenza di anomalie cardiache associate.
- Stadio I: ≥ 1,5 kg senza anomalie (95% sopravvivenza);
- Stadio II: < 1,5 kg o anomalie cardiache (55% sopravvivenza);
- Stadio III: < 1,5 kg e anomalie cardiache (22% sopravvivenza).
Classificazione basata sul gap tra i monconi
Oltre al peso alla nascita e alla presenza di malformazioni associate, il Gap tra i due monconi (la distanza) condiziona la prognosi per la possibilità di ricostruire tramite intervento chirurgico:
- Long gap: Gap inferiore 2,5 cm, buona anastomosi;
- Very long Gap: Gap tra 2,5 e 5,5 cm. Si deve trazionare molto l’esofago distale per una buona anastomosi (comunque a rischio di leakeage perché sotto tensione);
- Ultra long Gap: Gap > 5,5 cm costretti a intervento chirurgico in più tempi (soggetti con atresia di tipo I o II).

Malformazioni associate

I bambini con basso peso alla nascita hanno maggiori probabilità di avere altre anomalie dell’organogenesi. Più frequenti sono le anomalie muscolo-scheletriche, cardiovascolari, gastrointestinali e genitourinarie. Ciò è suggerito da prove genetiche, come spesso l’associazione con trisomia 18 (Sindrome di Edwards). La sindrome si manifesta con malformazioni congenite multiple in quasi tutti gli organi, ciclopia, malformazioni renali, difetti cardiaci strutturali (difetto del setto ventricolare, difetto interatriale, pervietà del dotto di Botallo), intestino sporgente al di fuori del corpo (onfalocele), atresia esofagea, ritardo mentale, ritardo nello sviluppo, deficit di crescita, difficoltà di alimentazione, difficoltà di respirazione, e artrogriposi (patologia muscolare che provoca contratture articolari multiple).
A volte sono malformazioni che interessano l’intestino primitivo con malformazioni ano-rettali che vanno dall’assenza dell’ano a sbocco dell’ano in posizione ectopica. La motilità dell’esofago è quasi sempre compromessa con peristalsi diminuita o assente soprattutto nella porzione distale.
Associata spesso è la tracheomalacia, che è una condizione o un incidente in cui la cartilagine che mantiene aperte le vie aeree (trachea) è morbida in modo tale che la trachea collassi parzialmente soprattutto durante l’aumento del flusso d’aria.
Altre volte sono associate in quadri sindromici ben precisi come:
- VACTERL: malformazioni Vertebrali, Ano imperforato (atresia anale) o ectopico, anomalie Cardiovascolari, fistola Tracheoesofagea, atresia dell’Esofago, anomalie Renali e Arti (Limbs).
- CHARGE: Coloboma di iride o retina; cardiopatie congenite (Heart), Atresia delle coane, Ritardo mentale e della crescita, Genitali ipoplasici o malformati, anomalie dell’orecchio e sordità (Ear).
Clinica
La clinica è caratterizzata da segni e sintomi tipici di un’occlusione. In particolar modo si avrà scialorrea (ipersalivazione) e disfagia dalla nascita. Se presente una fistola distale ci sarà irritazione chimica da acidità per cui c’è tosse, cianosi fino al distress respiratorio da polmonite chimica. Se presente fistola superiore la saliva può dare lo stesso effetto.
In caso di atresia non riconosciuta alla nascita si può avere polmoniti ab ingestis.
La Fistola ad H (senza atresia) sarà sicuramente misconosciuta e verrà diagnosticata anni dopo, in seguito a sintomatologia di distensione gastrica, difficoltà respiratorie, tosse e broncopolmoniti ricorrenti.

Diagnosi
In tutti i neonati, subito dopo il parto, si posiziona un sondino nasogastrico per valutare la pervietà dell’esofago. La normale lunghezza dell’esofago quando è pervio è 17 cm. Se si ferma a 9-13 cm è probabile ci sia atresia.
La diagnosi si deve sospettare:
- con la prova del sondino, da effettuare in tutti i nati;
- nel bambino sintomatico con ipersalivazione, tosse, cianosi, dispnea;
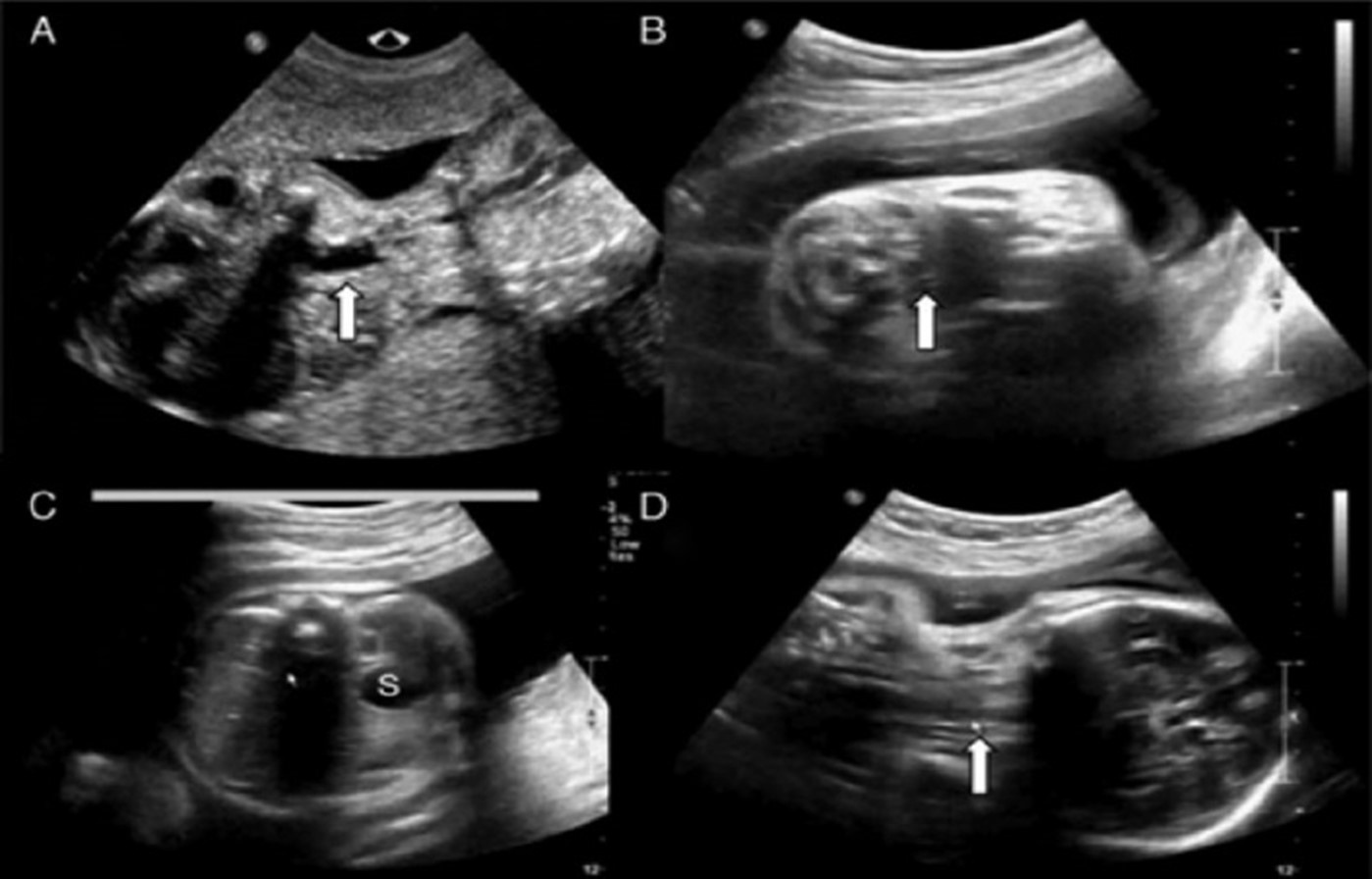
Assenza di moncone esofageo in sezione trasversale (B) e longitudinale (D).
Stomaco normale alla 33a settimana di gestazione (C).
con diagnosi ecografica prenatale (elementi di sospetto sono il Polidramnios e la mancata visualizzazione della bolla gastrica).
Fin dalla 11° settimana di gestazione il feto è in grado di deglutire una piccola quantità il liquido amniotico consentendo la visualizzazione dello stomaco (a partire dalla 24a settimana di gestazione). La presenza di AE determina l’arresto del turn-over di liquido amniotico con conseguente polidramnios.
- AE di I tipo: polidramnios costante e mancata visualizzazione della bolla gastrica.
- AE di III tipo: polidramnios incostante e normale visualizzazione della bolla gastrica se vi è il passaggio di liquido amniotico attraverso la FTE.
In tutti i bambini in cui non c’è avanzamento del sondino nasogastrico si fa un Rx torace-addome senza mdc che mostra la posizione in cui si è fermato il sondino. Se in regione toracica si fa diagnosi di atresia. Dall’Rx, inoltre, e dalla presenza di aria in addome, è possibile capire di che tipo è l’atresia e se c’è FTE. Se non c’è aria è di tipo 1 o tipo 2 (tutto opacato). Se è presente l’aria sarà tipo 3 o 4, perché passa nello stomaco dalla trachea per la presenza di una fistola.
Bisogna considerare tutte le alterazioni che possono essere associate e che ne impattano la prognosi; quindi, è importante effettuare un ECOCARDIO e un’ECOGRAFIA dei reni e delle vie urinarie.

Trattamento
Per decidere la strategia chirurgica basta sapere se c’è o meno una fistola esofagea distale. Se non c’è una fistola tracheoesofagea il gap è molto grande e questo preclude l’immediata correzione chirurgica (fistole ultra long gap). Si hanno due opzioni in tal caso: aspettare la crescita del bambino che permette allungamento del moncone superiore e dunque accorciamento dei gap con più facilità di operare (avviene circa in 12-16 settimane) oppure la sostituzione con colon o ileo.
Se invece c’è una fistola ed il paziente è in buone condizioni si può andare subito ad operare perché i monconi sono sufficientemente vicini.
In attesa dell’operazione in paziente con gap lungo a diagnosi fatta bisogna fare tre cose:
- porre una sonda di Replogle a due canali per aspirare saliva ed evitare l’ab ingestis;
- nutrizione Via Gastrostomia da preferire alla nutrizione parenterale (per evitare infezioni del catetere ed insufficienza epatica);
- copertura antibiotica profilattica.
La tecnica chirurgica utilizzata è quella della chiusura della fistola e anastomosi termino-terminale (anastomosi esofago-esofagea primaria) tra i monconi esofagei. Avviene con toracotomia o per via toracoscopia in alcuni centri, evitando con attenzione la vena azygos e il nervo vago ed esponendo la fistola per chiuderla con un punto. Sezionando l’esofago inferiore, si apre un occhiello nel moncone inferiore e lo si anastomizza con quello superiore (non è facile perché c’è differenza di calibro).
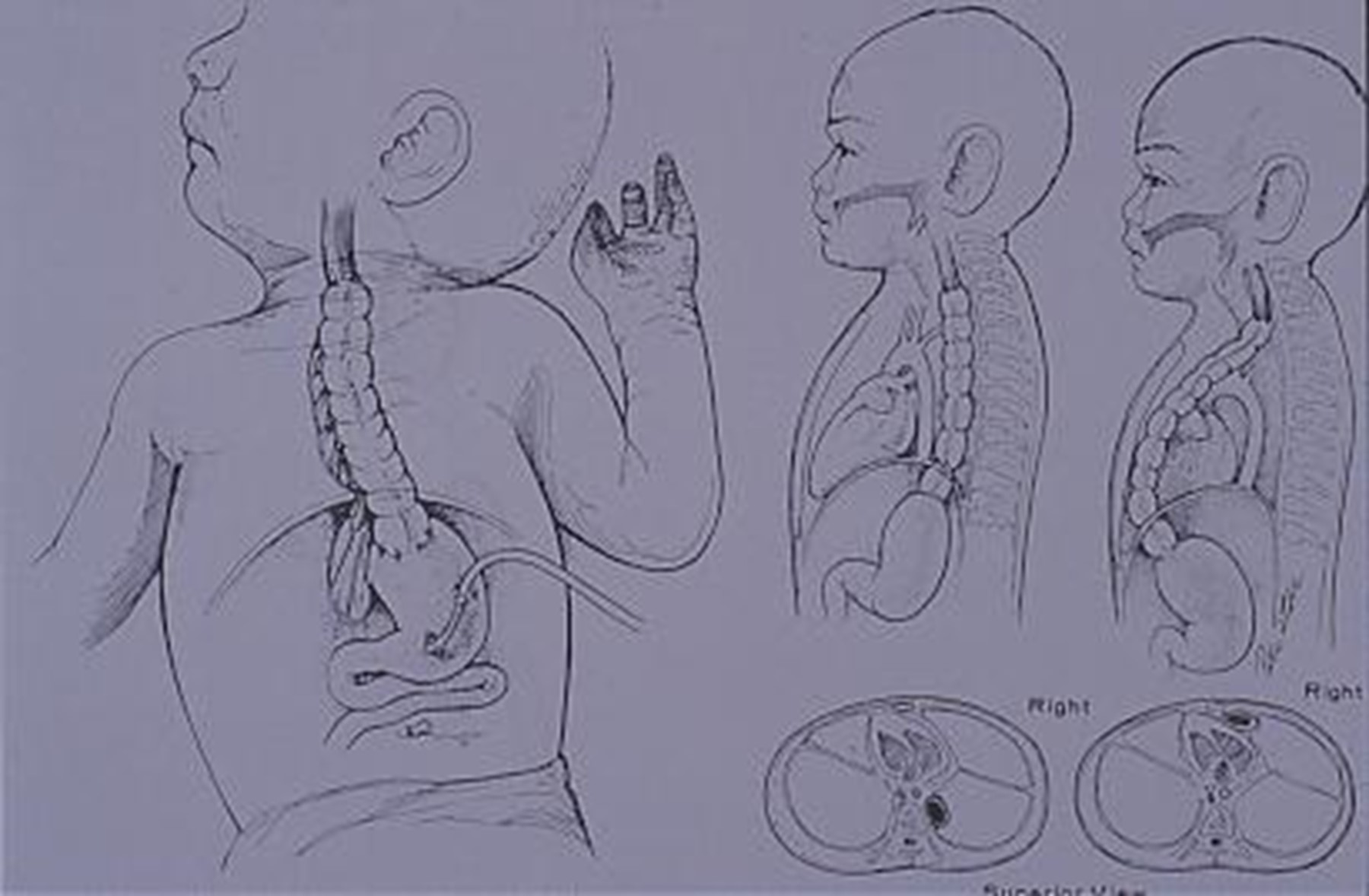
In soggetti in cui è molto difficile l’anastomosi si può usare colon, digiuno o ileo (esofagocolonplastica, come mostrato in figura), anche se è sempre meno usato.
Il controllo post-operatorio si esegue con Rx torace-addome.
COMPLICANZE POST-OPERATORIE PRECOCI:
- Deiscenza dell’anastomosi in tensione e filtrazione di saliva nel mediastino (evitata con drenaggio in mediastino);
- Stenosi nel punto dell’anastomosi (trattate con dilatazione pneumatica);
- Lesione del dotto toracico con chilotorache (drenaggio del chilo per mesi interi).
COMPLICANZE TARDIVE:
- Disfagia iatrogena per disturbi della deglutizione oppure da stenosi;
- Giunzione esofago-gastrica incontinente per alterata motilità esofagea e reflusso gastroesofageo;
- In pazienti con deiscenza formazione di fistola tra anastomosi e trachea;
- Tracheomalacia per indebolimento della trachea membranosa residuata dalla fistola.
Fonte: Chirurgia pediatrica.