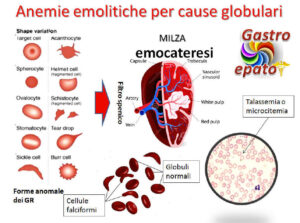
Le anemie emolitiche congenite sono condizioni in cui i globuli rossi hanno una vita più breve del normale a causa di difetti intrinseci nel loro metabolismo. Tra queste, le enzimopatie giocano un ruolo importante, poiché la carenza di alcuni enzimi fondamentali rende gli eritrociti più vulnerabili alla distruzione.
- Alterazioni della via della glicolisi aerobica (deficit di glucosio-6-fosfato deidrogenasi)
- Alterazioni della via della glicolisi anaerobica (carenza di piruvato-chinasi)
- Alterazioni del metabolismo dei nucleotidi (carenza di pirimidina-5′-nucleotidasi ed eccesso di adenosina deaminasi)
Alterazioni della via della glicolisi aerobica (deficit di glucosio-6-fosfato deidrogenasi)
Uno dei deficit più comuni è quello della glucosio-6-fosfato deidrogenasi (G6PD), un enzima cruciale per proteggere le cellule dallo stress ossidativo. Questa condizione è particolarmente diffusa nelle aree in cui la malaria è endemica, come il Mediterraneo, l’Africa e il Sud-Est asiatico. Il motivo? Si pensa che la carenza di G6PD possa offrire una certa resistenza al parassita della malaria, spiegando perché questa mutazione sia stata favorita dall’evoluzione in queste regioni.
La particolarità di questo deficit è che si tratta di una condizione legata al cromosoma X (Xq28). Questo significa che i maschi, avendo un solo cromosoma X, sono più esposti: se ereditano il gene difettoso, manifesteranno la malattia. Le donne, invece, avendo due cromosomi X, sono spesso portatrici sane, perché il cromosoma sano può compensare quello alterato a causa dell’inattivazione casuale del cromosoma X (lionizzazione). Tuttavia, in alcuni casi (ad esempio se entrambi i cromosomi X sono mutati o per un fenomeno di inattivazione sbilanciata), anche le donne possono sviluppare sintomi.
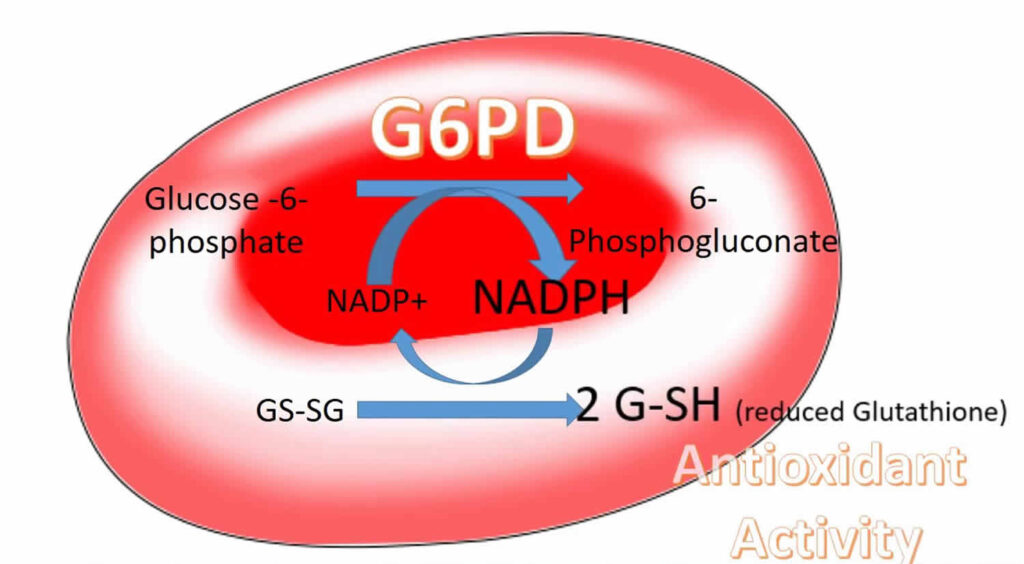
La G6PD è un enzima chiave nella via dei pentosi-fosfati, una via metabolica che produce NADPH, una molecola essenziale per neutralizzare i radicali liberi. Senza un adeguato NADPH, il glutatione (un importante antiossidante) non può funzionare correttamente, e l’emoglobina diventa vulnerabile all’ossidazione. Quando questo accade, si formano degli aggregati chiamati corpi di Heinz, che danneggiano la membrana dei globuli rossi, rendendoli rigidi e facili alla distruzione con conseguente emolisi intravascolare.
La maggior parte delle persone con deficit di G6PD vive senza sintomi, almeno finché non entra in contatto con qualcosa che scatena una crisi emolitica. Questi fattori scatenanti sono solitamente sostanze ossidanti, come alcuni farmaci (FANS, sulfamidici, antimalarici), chetoacidosi, infezioni (soprattutto polmoniti o epatiti virali) o persino l’ingestione di fave – una condizione nota come favismo.
Quando si verifica una crisi emolitica intravascolare, i sintomi compaiono rapidamente: stanchezza, pallore, ittero (pelle e occhi gialli) e urine scure (per la presenza di emoglobinuria). Nei casi più gravi, può esserci anche un rischio di insufficienza renale acuta a causa del carico di emoglobina filtrata dai reni.
Più raramente si manifesta con una sindrome emolitica cronica con splenomegalia.
Il sospetto diagnostico nasce spesso dall’anamnesi: un paziente che, dopo aver assunto un farmaco o mangiato fave, sviluppa improvvisamente anemia e ittero. Per confermare la diagnosi, si misura l’attività dell’enzima G6PD nel sangue, la presenza di emazie con inclusioni o corpi di Heinz
Attenzione, però: durante una crisi emolitica, i globuli rossi giovani (reticolociti) hanno ancora un po’ di attività enzimatica residua, quindi il test potrebbe risultare falsamente normale. Per questo, è meglio ripeterlo a distanza di qualche settimana dall’episodio acuto.

Non esiste una cura definitiva per il deficit di G6PD, ma la strategia principale è prevenire le crisi. Chi ne soffre deve evitare farmaci ossidanti (lista dettagliata da fornire al paziente) e alimenti a rischio (come le fave).
In caso di crisi emolitica è importante l’idratazione e monitoraggio della funzionalità renale, a volte è necessaria una trasfusione di sangue, soprattutto se l’anemia è grave. La splenectomia è utile solo in un numero limitato di casi.
Nei neonati, questo deficit può causare ittero neonatale severo, che richiede fototerapia o, nei casi più gravi, exsanguinotrasfusione per evitare danni cerebrali. Per questo, in alcune regioni ad alta prevalenza, lo screening per il deficit di G6PD viene fatto alla nascita.
Nella maggior parte dei casi, la prognosi è buona con un’adeguata gestione.
Le crisi emolitiche sono autolimitanti, poiché gli eritrociti più vecchi (deficitari) vengono eliminati, mentre i reticolociti (con maggiore attività G6PD residua) sopravvivono.
Now loading…
Alterazioni della via della glicolisi anaerobica (carenza di piruvato-chinasi)
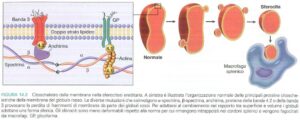
Il deficit di piruvato chinasi rappresenta la forma più comune tra queste enzimopatie. Si tratta di una condizione ereditaria che segue un pattern autosomico recessivo, il che significa che entrambi i genitori devono essere portatori del gene alterato affinché il figlio manifesti la malattia. Ciò che rende particolare questo deficit è il suo impatto sul metabolismo energetico dell’eritrocita: la piruvato chinasi è infatti l’enzima chiave che permette la produzione di ATP attraverso la glicolisi anaerobica. Senza un adeguato apporto energetico, i globuli rossi diventano rigidi e fragili, venendo eliminati precocemente dal sistema reticolo-endoteliale, in particolare dalla milza.
Il quadro clinico che ne risulta è piuttosto caratteristico. I pazienti sviluppano un’anemia emolitica cronica che può manifestarsi già nei primi mesi di vita, con ittero intermittente (dovuto all’aumento della bilirubina libera) e splenomegalia marcata. A differenza del favismo, dove le crisi emolitiche sono scatenate da fattori esterni, in questo caso l’emolisi è costante, anche se può peggiorare in situazioni di stress metabolico come infezioni o gravidanza.
Nei bambini, l’anemia cronica può portare a ritardi di crescita, mentre negli adulti si osserva spesso una buona compensazione, almeno fino a quando la milza non raggiunge dimensioni tali da causare sintomi meccanici.

La diagnosi di questa condizione richiede un approccio articolato. L’aspetto più importante è la misurazione dell’attività enzimatica, che però può essere influenzata dalla presenza di reticolociti (che possiedono una maggiore attività residua). Per questo motivo, a volte è necessario ricorrere a test genetici per identificare le mutazioni responsabili.
All’esame dello striscio periferico, i globuli rossi possono apparire normali o mostrare solo lievi alterazioni morfologiche, rendendo spesso necessario escludere altre cause di emolisi cronica.
Gli esami di laboratorio mostrano segni di emolisi (aumento della bilirubina indiretta, LDH elevato, aptoglobina bassa).
Per quanto riguarda il trattamento, le opzioni sono purtroppo limitate. La terapia si basa principalmente sulla supplementazione con acido folico per sostenere l’eritropoiesi riducendo crisi megaloblastiche e, nei casi più gravi, su trasfusioni periodiche.
La splenectomia può offrire un miglioramento in alcuni pazienti, riducendo il grado di emolisi, ma raramente risolve completamente il problema. Recentemente, sono stati fatti progressi nel trapianto di midollo osseo, che rappresenta l’unica opzione potenzialmente curativa, anche se riservata ai casi più severi.
Caricamento…
Alterazioni del metabolismo dei nucleotidi (carenza di pirimidina-5′-nucleotidasi ed eccesso di adenosina deaminasi)
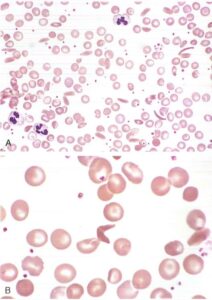
Passando al metabolismo dei nucleotidi, due condizioni meritano particolare attenzione. La prima è la carenza di pirimidina-5′-nucleotidasi, un enzima coinvolto nella degradazione dell’RNA eritrocitario. In sua assenza, si accumulano prodotti di degradazione che formano inclusioni visibili al microscopio come punteggiatura basofila. Questo reperto, però, non è specifico e può essere riscontrato anche in altre condizioni come le talassemie o l’intossicazione da piombo, rendendo necessaria una diagnosi differenziale accurata.
L’eccesso di adenosina deaminasi, invece, rappresenta una condizione più rara ma ugualmente interessante. Questo enzima, normalmente presente in piccole quantità, quando prodotto in eccesso altera il delicato equilibrio dei nucleotidi eritrocitari, portando a una ridotta sintesi di ATP e quindi a una minore sopravvivenza dei globuli rossi. Il quadro clinico è simile a quello di altre anemie emolitiche congenite, ma la diagnosi richiede test enzimatici specifici.

Queste condizioni, seppur rare, insegnano molto sul complesso metabolismo dell’eritrocita e sulle sue vulnerabilità. Mostrano come alterazioni in vie metaboliche apparentemente secondarie possano avere ripercussioni cliniche significative. Inoltre, pongono sfide diagnostiche non banali, dove l’integrazione tra dati di laboratorio, osservazione morfologica e test genetici diventa essenziale per arrivare a una diagnosi corretta.
La ricerca in questo campo sta facendo progressi interessanti, con lo sviluppo di nuove tecniche diagnostiche e l’esplorazione di terapie geniche che potrebbero, in futuro, offrire soluzioni più definitive rispetto alle attuali opzioni terapeutiche. Nel frattempo, la gestione di questi pazienti richiede un approccio multidisciplinare, attento non solo agli aspetti ematologici ma anche alle possibili complicanze a lungo termine, come i danni d’organo da emocromatosi secondaria o le conseguenze della splenomegalia massiva.
Caricamento….