La distrofia muscolare di Duchenne (DMD) rappresenta il paradigma della complessità nelle malattie rare, configurandosi come la forma più severa di distrofinopatia nell’infanzia.
Caratterizzata da una degenerazione progressiva e inesorabile della muscolatura scheletrica, liscia e cardiaca, la DMD trae origine da un difetto genetico nel locus Xp21.2 che impedisce la sintesi della distrofina, una proteina citoscheletrica essenziale per l’integrità del sarcolemma.12

La sua prevalenza globale, stimata tra 1 su 3.500 e 1 su 5.000 nati vivi maschi, la colloca tra le sfide prioritarie della neurologia pediatrica e della medicina genetica contemporanea.3
Storicamente descritta nel XIX secolo da Guillaume Benjamin Amand Duchenne, la comprensione della patologia ha attraversato tre ere fondamentali:
- l’era clinica, focalizzata sulla descrizione dei sintomi e della progressione motoria;
- l’era genetica, inaugurata nel 1986 con l’identificazione del gene DMD;
- l’era terapeutica attuale, caratterizzata dal tentativo di correggere il difetto molecolare attraverso strategie di precisione.4
L’evoluzione della gestione clinica, passata dalla mera assistenza palliativa a un approccio multidisciplinare standardizzato, ha esteso significativamente l’aspettativa di vita, portando a una nuova popolazione di pazienti adulti che affrontano sfide sistemiche precedentemente inesplorate.56

Eziologia molecolare e architettura genomica del locus DMD

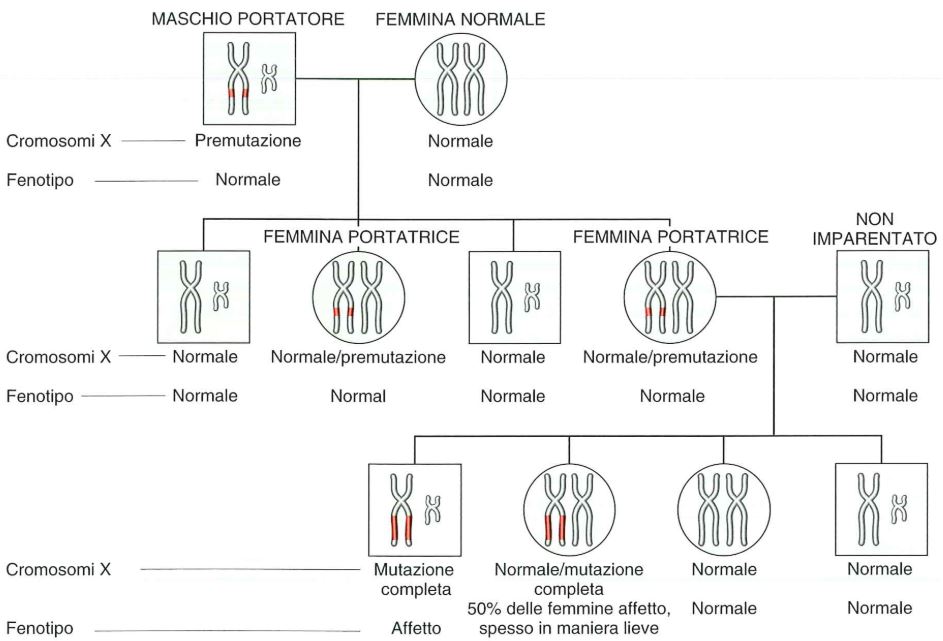
L’eziologia della DMD risiede in mutazioni patogenetiche a carico del gene DMD, situato sul cromosoma X. Questo gene detiene il primato per dimensioni nel genoma umano, estendendosi per circa 2,4 milioni di basi e comprendendo 79 esoni.7
La straordinaria ampiezza del gene non è solo una curiosità biologica, ma una determinante eziologica fondamentale: la sua lunghezza lo rende intrinsecamente vulnerabile a un elevato tasso di mutazioni spontanee e de novo, che rappresentano circa il 30% dei casi diagnosticati, mentre il restante 70% segue un modello di eredità recessiva legata all’X.89
Le mutazioni che colpiscono il gene DMD mostrano una distribuzione non casuale, concentrandosi in specifiche regioni denominate hotspot. Circa il 60-70% dei pazienti presenta delezioni di uno o più esoni, localizzate prevalentemente nel dominio centrale (esoni 44-55) o nella regione N-terminale (esoni 2-20).
Le duplicazioni esoniche interessano circa il 10% dei casi, mentre le piccole mutazioni puntiformi, incluse le mutazioni nonsense, rappresentano il 15-30% dello spettro mutazionale.10
Trascritti e isoforme: la complessità tessuto-specifica
Il gene DMD non codifica per una singola proteina, ma attraverso un sofisticato sistema di promotori interni e splicing alternativo, genera molteplici isoforme espresse in tessuti diversi. L’isoforma full-length di 427 kDa (Dp427) è tipica dei muscoli striati e dei neuroni corticali.11

Tuttavia, la presenza di isoforme corte come Dp140, Dp116 e Dp71 spiega il coinvolgimento di organi extra-muscolari. Ad esempio, la mancanza di isoforme specifiche nel sistema nervoso centrale è correlata alle manifestazioni cognitive e neuropsichiatriche, suggerendo che la distrofina svolga un ruolo cruciale non solo come ancora strutturale, ma anche nella stabilizzazione delle sinapsi e nella modulazione dei segnali neuronali.
| Caratteristica genomica | Dettaglio tecnico | Implicazione clinica |
|---|---|---|
| Localizzazione | Cromosoma Xp21.2 | Trasmissione recessiva legata all’X |
| Dimensione Gene | 2,4 Megabasi (79 esoni) | Elevata frequenza di mutazioni de novo |
| Principale Mutazione | Delezioni esoniche (65%) | Bersaglio per terapie di exon skipping |
| Mutazioni Nonsense | ~10-13% dei casi | Eleggibilità per trattamento con Ataluren |
| Isoforme Brevi | Dp140, Dp71, Dp116 | Coinvolgimento cognitivo e retinico |
Caricamento…
Fisiopatogenesi: la cascata della degenerazione muscolare
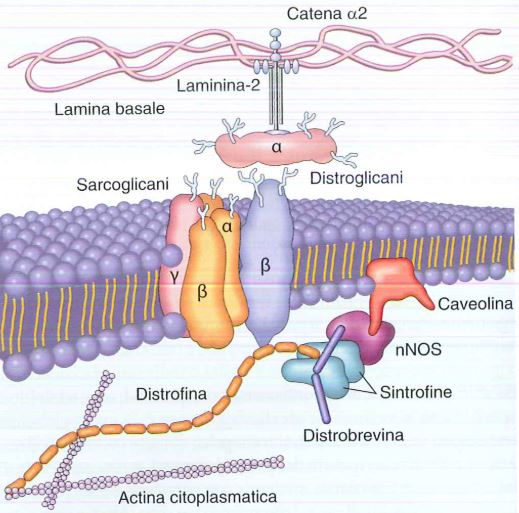
La fisiopatogenesi della DMD non può essere ridotta a una semplice debolezza strutturale; essa consiste in una cascata di eventi biochimici e cellulari che trasformano una stabilità di membrana compromessa in un’infiammazione cronica e fibrosi sistemica. La distrofina funge da collegamento meccanico vitale tra l’actina del citoscheletro interno e la matrice extracellulare attraverso il complesso glicoproteico associato alla distrofina (DGC), che include distroglicani, sarcoglicani, sintrofine e distrobrevine.
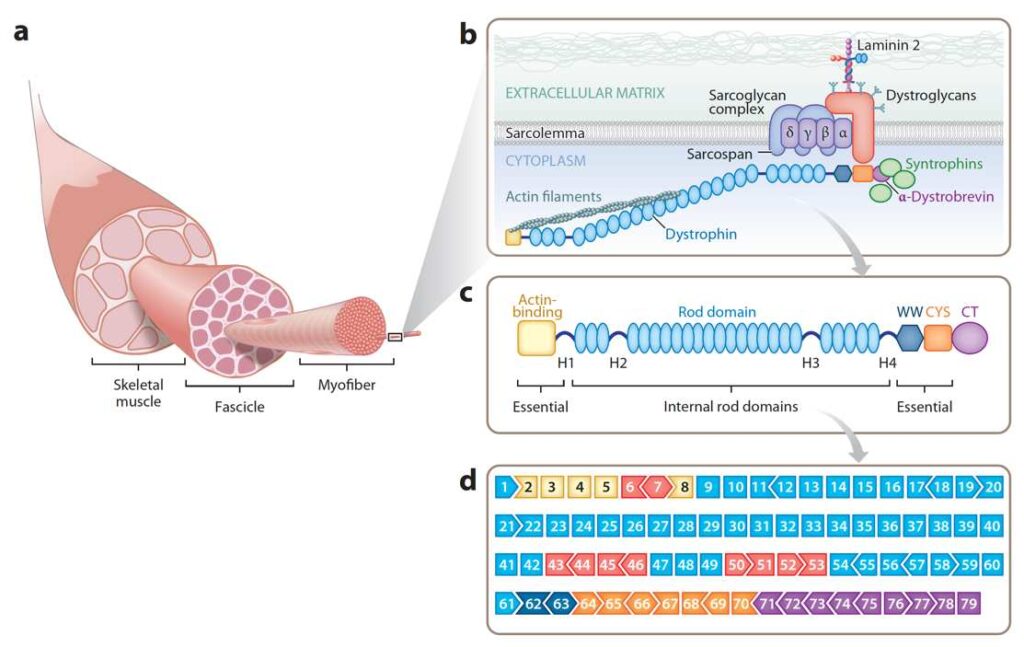
a) Rappresentazione del muscolo scheletrico, costituito da miofibre. Ogni miofibra contiene l’unità funzionale del muscolo detto sarcomero.
b) Complesso Distrofina-Glicoproteine (DGC): illustrazione delle interazioni della distrofina con i filamenti di actina e le proteine del DGC nel sarcolemma.
c) Struttura proteica della distrofina: la parte N terminale contiene il dominio legante l’actina, mentre nella parte C terminale sono presenti i siti di legame alla sintrofina, distrobrevina e distroglicano. Il dominio centrale agisce come una molla elastica tra le due estremità.
d) Struttura a 79 esoni del gene DMD; il codice a colori identifica in arancione le mutazioni più frequenti.12
In assenza di questa proteina, il sarcolemma diventa fragile e suscettibile a lacerazioni durante i normali cicli di contrazione e rilassamento. Il modello dell'”esplosione cellulare” descrive accuratamente come la perdita dell’integrità della membrana porti a un aumento della pressione interna e alla successiva morte della fibra muscolare.13
Omeostasi del calcio e stress ossidativo mitocondriale

Uno dei primi eventi patologici è la perdita della permeabilità selettiva del sarcolemma. L’afflusso massiccio di ioni calcio (Ca2+) nello spazio intracellulare attiva le calpaine, enzimi proteolitici che iniziano la degradazione indiscriminata delle proteine contrattili.14
Questo sovraccarico di calcio altera anche la funzione mitocondriale; i mitocondri, nel tentativo di sequestrare l’eccesso di calcio, subiscono un collasso del potenziale di membrana, riducendo la produzione di ATP e aumentando drasticamente la generazione di specie reattive dell’ossigeno (ROS). La disfunzione mitocondriale è ulteriormente aggravata dal fallimento dei processi di mitofagia, portando all’accumulo di organelli danneggiati che alimentano lo stress ossidativo e la necrosi cellulare.
Risposta infiammatoria e sostituzione fibroadiposa
La necrosi delle fibre muscolari rilascia nel torrente circolatorio molecole pro-infiammatorie e marker enzimatici come la creatin-chinasi (CK). Questo attiva una risposta immunitaria cronica mediata dalla via di segnalazione NF-κB, che promuove l’infiltrazione di macrofagi e cellule T nel tessuto muscolare.
Sebbene inizialmente la riparazione sia tentata dalle cellule satelliti (le cellule staminali del muscolo), lo stimolo rigenerativo continuo porta all’esaurimento della loro capacità proliferativa.15
L’ambiente cronico infiammatorio favorisce l’attivazione dei fibroblasti, che sostituiscono il tessuto contrattile con cicatrici fibrose e tessuto adiposo, un processo che clinicamente si manifesta con la perdita di forza e la pseudoipertrofia.

Classificazione delle distrofinopatie
Le distrofinopatie comprendono un ampio spettro di gravità clinica, unificate dall’eziologia genetica ma distinte dalla funzionalità della proteina prodotta.
Distrofia Muscolare di Duchenne (DMD)
La DMD è la forma più grave, solitamente causata da mutazioni out-of-frame. In questi casi, la delezione o duplicazione altera il modulo di lettura della tripletta di basi, portando alla formazione di un codone di stop prematuro e alla degradazione del messaggero o alla produzione di una proteina non funzionale e instabile
Il livello di distrofina residua è generalmente inferiore al 5% della norma.
Distrofia Muscolare di Becker (BMD)
La BMD rappresenta la variante più lieve, spesso derivante da mutazioni in-frame. Lo schema di lettura viene preservato, consentendo la traduzione di una proteina accorciata ma parzialmente funzionale.16
I livelli di distrofina nella BMD variano dal 20% all’80%, determinando un esordio più tardivo e una progressione significativamente più lenta rispetto alla DMD.
Varianti atipiche
È fondamentale notare che la “reading frame rule” non è assoluta e presenta eccezioni in circa il 10% dei casi. Ad esempio, alcune delezioni in-frame nella regione N-terminale (come la delezione degli esoni 3-7) possono causare un fenotipo severo tipo Duchenne, mentre mutazioni out-of-frame possono talvolta manifestarsi come Becker a causa di meccanismi di skipping esonico spontaneo che ripristinano parzialmente il modulo di lettura.
Altre forme includono la cardiomiopatia dilatativa isolata legata all’X (XLDCM), dove il danno muscolare scheletrico è minimo o assente, ma il miocardio è gravemente colpito a causa di mutazioni che influenzano i trascritti cardiaci specifici.
| Fenotipo | Mutazione tipica | Funzione Distrofina | Esordio clinico |
|---|---|---|---|
| DMD | Out-of-frame | Praticamente nulla (<5%) | 2-5 anni |
| BMD | In-frame | Ridotta o accorciata (20-80%) | Adolescenza/Età adulta |
| DMD Intermedia | Variabile | Livelli borderline | 5-10 anni |
| XLDCM | Specifica cardiaca | Deficit cardiaco predominante | 2ª-4ª decade |
Storia naturale e progressione dei quadri clinici
La DMD segue un decorso evolutivo che può essere categorizzato in stadi funzionali definiti, riflettendo la progressiva perdita di tessuto muscolare e l’insufficienza di vari sistemi d’organo.
Fase presintomatica e diagnosi precoce (0-4 anni)
Sebbene i pazienti nascano con elevati livelli di CK, i sintomi motori sono spesso assenti o minimi nei primi due anni di vita. I primi segnali possono includere un lieve ritardo nel controllo della testa e del collo, o una deambulazione iniziata dopo i 18 mesi.
È interessante notare che in questa fase possono essere già presenti ritardi nel linguaggio e disturbi del neurosviluppo come l’autismo o l’ADHD, spesso prima che emerga la debolezza muscolare manifesta.
Stadio deambulante precoce (5-7 anni)
In questo periodo, i segni classici diventano evidenti. I bambini presentano un’andatura anserina con base allargata e una marcata iperlordosi lombare, dovuta alla debolezza dei muscoli stabilizzatori del cingolo pelvico e del tronco. La difficoltà a salire le scale e le cadute frequenti sono comuni.17
Il segno di Gowers, l’uso delle braccia per “arrampicarsi” sulle proprie gambe per alzarsi da terra, è un indicatore patognomonico della debolezza dei muscoli prossimali degli arti inferiori.
La pseudoipertrofia dei polpacci raggiunge il suo massimo vigore clinico in questa fase.

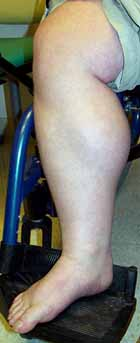
Nelle fasi precoci la DMD colpisce i muscoli delle spalle e degli arti superiori e delle cosce. Questo comporta difficoltà nell'alzarsi da terra, salire le scale, mantenere l'equilibrio e alzare le braccia.
Caricamento....
Stadio deambulante tardivo e transizione (8-12 anni)
La debolezza progredisce rapidamente verso i muscoli distali e gli arti superiori. La capacità di camminare autonomamente declina, con un’età media di perdita della deambulazione fissata a 9,5 anni nei pazienti non trattati con steroidi e a 12-13 anni in quelli trattati. Durante questa fase di transizione, i pazienti possono necessitare di una carrozzina per lunghe distanze prima di perdere completamente la capacità di stare in piedi.
Fase non deambulante e complicanze sistemiche (adolescenza ed età adulta)
Now loading…
Una volta persa la deambulazione, la scoliosi tende a svilupparsi rapidamente a causa dello squilibrio muscolare paravertebrale, compromettendo ulteriormente la già ridotta capacità respiratoria.18
- Insufficienza respiratoria: l’indebolimento del diaframma e dei muscoli intercostali porta a una ventilazione restrittiva. L’accumulo di secrezioni e la ridotta efficacia della tosse predispongono ad atelettasie e infezioni polmonari ricorrenti.
- Cardiomiopatia: il muscolo cardiaco subisce processi di fibrosi simili a quelli del muscolo scheletrico. Entro i 18 anni, circa il 50% dei pazienti presenta segni di scompenso cardiaco, che progredisce verso la cardiomiopatia dilatativa globale e aritmie potenzialmente letali.19
- Coinvolgimento gastrointestinale: possono insorgere difficoltà nella deglutizione (disfagia), reflusso gastroesofageo e stipsi cronica per ipomotilità della muscolatura liscia.


Protocolli diagnostici
La diagnosi tempestiva è cruciale per massimizzare i benefici dei trattamenti modificanti la malattia. Il percorso diagnostico moderno si è spostato significativamente verso metodi molecolari non invasivi.
Screening biochimico iniziale
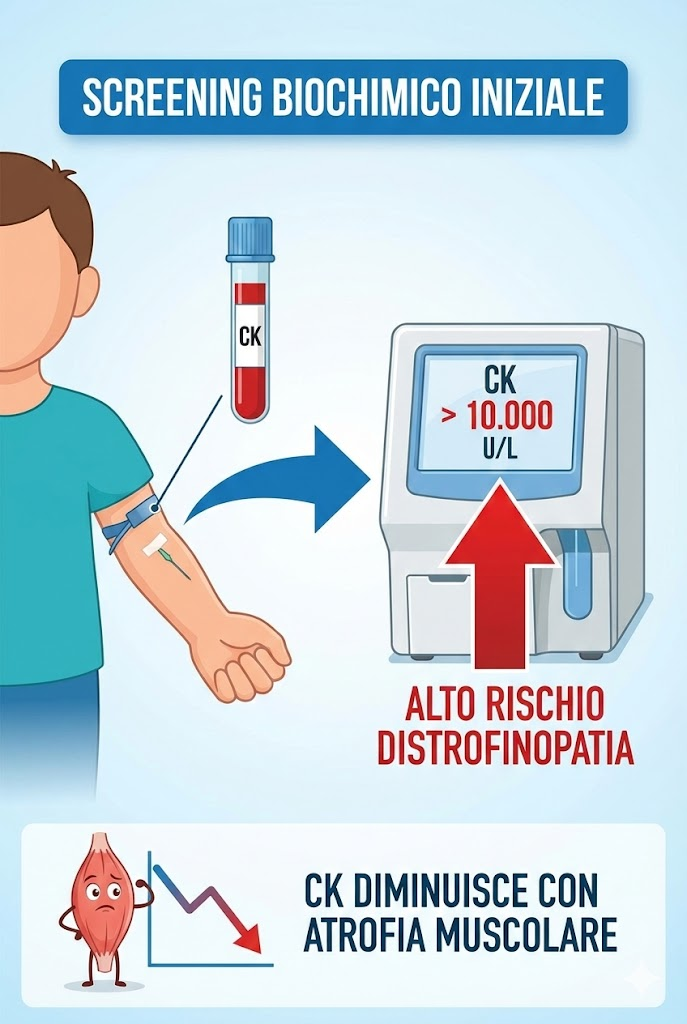
Il sospetto clinico deve essere immediatamente seguito dal dosaggio della creatin-chinasi (CK). Valori superiori a 10-20 volte il limite normale nei bambini maschi indicano fortemente una distrofinopatia; nella DMD classica, i livelli possono superare i 10.000-20.000 U/L.
Sebbene elevati, i livelli di CK tendono a diminuire nelle fasi terminali della malattia a causa dell’esaurimento della massa muscolare.
Un aumento cospicuo e persistente dei livelli di CPK è indicativo di una patologia muscolare ma non necessariamente di Distrofia Muscolare di Duchenne.
Gold Standard genetico
Now loading…
La diagnosi definitiva richiede l’identificazione della mutazione nel gene DMD.
- MLPA (Multiplex Ligation-dependent Probe Amplification): questa tecnica è il primo passo per individuare delezioni e duplicazioni esoniche, responsabili della vasta maggioranza dei casi.
- Sequenziamento dell’intero gene (NGS o Sanger): necessario per individuare mutazioni puntiformi, mutazioni nonsense e piccoli riarrangiamenti non rilevabili con la MLPA.
La diagnosi prenatale è possibile nelle famiglie con una diagnosi confermata mediante analisi molecolari.
Biopsia muscolare e immunoistochimica
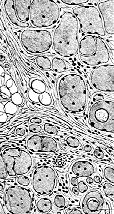
La biopsia muscolare è oggi raccomandata solo se i test genetici non sono conclusivi o per studi di ricerca. L’analisi morfologica mostra una variabilità del diametro delle fibre, necrosi, rigenerazione delle miofibre e un aumento del tessuto connettivo endomisiale con sostituzione del tessuto muscolare in tessuto adiposo.
L’immunofluorescenza per la distrofina rivela la sua completa assenza nel sarcolemma nella DMD, mentre mostra una colorazione ridotta o a mosaico nella BMD e nelle femmine portatrici sintomatiche.
Caricamento…
Strategie terapeutiche
Sebbene non esista ancora una cura risolutiva, la gestione farmacologica si è evoluta verso un approccio combinato che mira a rallentare la degenerazione e correggere il difetto genetico sottostante.
Farmacoterapia con corticosteroidi
I corticosteroidi rimangono il pilastro del trattamento.
Prednisone (0,75 mg/kg/die) e Deflazacort (0,9 mg/kg/die) stabilizzano la funzione motoria e polmonare, ritardando la perdita della deambulazione e riducendo la necessità di chirurgia per la scoliosi.
Tuttavia, i loro benefici devono essere bilanciati contro effetti collaterali come l’osteoporosi, il ritardo della crescita, l’aumento di peso e le cataratte.
Terapie a Bersaglio Genico e Precision Medicine
L’introduzione di terapie mutazione-specifiche ha segnato l’inizio della medicina personalizzata per la DMD.

- Ataluren (Translarna): progettato per mutazioni nonsense, agisce “nascondendo” il codone di stop prematuro per permettere la traduzione della proteina intera.20
Nonostante l’approvazione condizionata iniziale, nel 2024 l’EMA ha confermato la raccomandazione di non rinnovare l’autorizzazione a causa della mancanza di prove definitive di efficacia negli studi clinici più recenti. - Exon skipping: oligonucleotidi antisenso come Eteplirsen (esone 51), Golodirsen (esone 53) e Casimersen (esone 45) mirano a trasformare la DMD in un fenotipo tipo Becker. Sebbene approvati negli Stati Uniti dalla FDA, l’EMA è rimasta cauta, richiedendo dati clinici più robusti sul miglioramento funzionale a lungo termine oltre al semplice aumento dei livelli di distrofina nella biopsia.
- Terapia Genica con microdistrofina: Elevidys (delandistrogene moxeparvovec) introduce una versione accorciata ma funzionale del gene DMD tramite un vettore virale AAV.2122
Nonostante il successo regolatorio negli USA, l’EMA ha respinto la richiesta di autorizzazione nel luglio 2025, citando il mancato raggiungimento della significatività statistica nei test motori (NSAA) e gravi preoccupazioni per la sicurezza legate a casi di insufficienza epatica acuta.23
Inibitori delle Istone Deacetilasi (HDAC): Givinostat
Il Givinostat rappresenta una nuova classe di farmaci non steroidei. Agisce modulando l’attività delle HDAC, che nei muscoli distrofici è deregolata, favorendo un ambiente pro-rigenerativo e contrastando la fibrosi.2425
I risultati dello studio EPIDYS hanno mostrato un rallentamento significativo nel declino motorio (test dei 4 gradini), portando l’EMA a raccomandare l’approvazione condizionata nell’aprile 2025 per pazienti deambulanti dai 6 anni in su.
| Farmaco | Meccanismo d’azione | Stato approvativo (2024-2025) | Note cliniche |
|---|---|---|---|
| Prednisone | Glucocorticoide standard | Approvato / Standard di cura | Effetti sistemici significativi |
| Vamorolone | Corticosteroide dissociativo | Approvato EMA (Dicembre 2023) | Minori effetti su crescita e ossa |
| Ataluren | Read-through mutazioni nonsense | Parere negativo EMA (Ottobre 2024) | Efficacia non confermata in fase 3 |
| Givinostat | Inibitore HDAC | Raccomandazione EMA (Aprile 2025) | Riduce fibrosi e infiammazione |
| Elevidys | Terapia Genica (Microdistrofina) | Rifiutato EMA (Luglio 2025) | Approvato negli USA (FDA) |
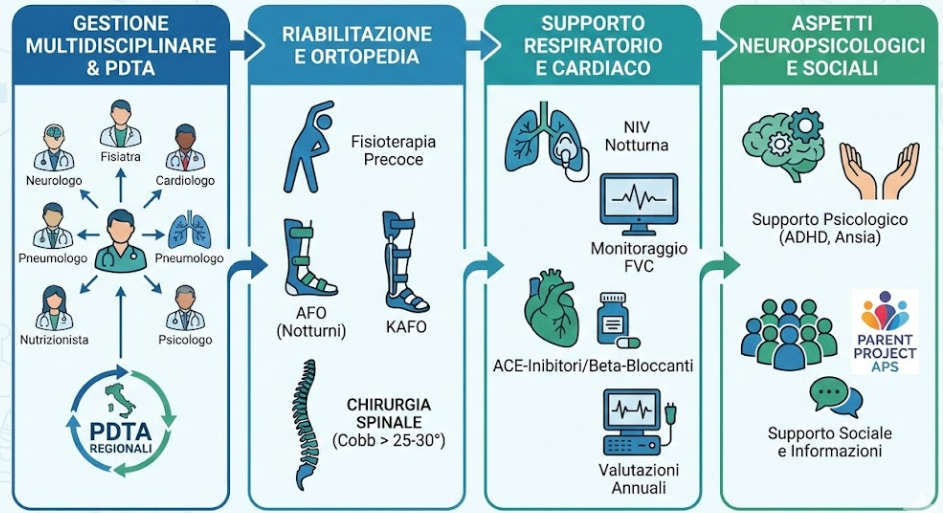
Gestione Multidisciplinare e PDTA in Italia
In Italia, la presa in carico dei pazienti con DMD è coordinata attraverso i Percorsi Diagnostico Terapeutici Assistenziali (PDTA) regionali, che riflettono le linee guida internazionali.
La gestione multidisciplinare coinvolge neurologi, fisiatri, cardiologi, pneumologi, nutrizionisti e psicologi.
Riabilitazione e ortopedia
La fisioterapia deve iniziare precocemente per mantenere l’estensibilità muscolare e prevenire le contratture, specialmente a carico delle caviglie e delle ginocchia.
L’uso di ortesi notturne tipo AFO è raccomandato per contrastare l’equinismo del piede, mentre i tutori lunghi KAFO possono aiutare a prolungare il tempo di stazione eretta.
La chirurgia spinale per la scoliosi è considerata quando l’angolo di Cobb supera i 25-30 gradi per preservare la funzione respiratoria.
Supporto respiratorio e cardiaco
Il monitoraggio della funzionalità polmonare (FVC, picco di flusso di tosse) deve essere effettuato ogni 6 mesi dopo la perdita della deambulazione.
La ventilazione meccanica non invasiva (NIV) notturna è indicata per l’insufficienza respiratoria, migliorando significativamente la qualità del sonno e la sopravvivenza a lungo termine.
Dal punto di vista cardiaco, l’uso profilattico di ACE-inibitori o beta-bloccanti è oggi lo standard di cura per rallentare l’insorgenza della cardiomiopatia dilatativa, con valutazioni strumentali annuali (ECG ed ecocardiogramma/RM cardiaca).
Aspetti neuropsicologici e sociali
Circa il 30% dei pazienti presenta disabilità intellettiva, spesso con una discrepanza tra quoziente intellettivo verbale e di performance.
La gestione di ADHD, ansia e disturbi dello spettro autistico richiede un supporto psicologico costante per il paziente e la famiglia.
Le associazioni come Parent Project aps sono fondamentali per la rete di supporto sociale e per la diffusione di informazioni aggiornate sulle sperimentazioni cliniche in Italia.2627

Prognosi e prospettive future
La prognosi della DMD è radicalmente mutata negli ultimi trent’anni. Grazie agli interventi multidisciplinari, l’aspettativa di vita media si è spostata dai 19-20 anni ai 30-35 anni, con molti pazienti che raggiungono l’età adulta e partecipano attivamente alla vita sociale e accademica.
Now loading…
Le direzioni future della ricerca includono:
- gene editing (CRISPR/Cas9): tentativi di correzione definitiva del gene nel tessuto muscolare, superando i limiti dei vettori virali e della microdistrofina;
- inibitori della miostatina: farmaci mirati ad aumentare la massa muscolare neutralizzando il regolatore negativo della crescita muscolare;
- terapie anti-fibrotiche e rigenerative: sviluppo di molecole che agiscono sulla nicchia delle cellule staminali muscolari per preservare la capacità di riparazione;
- ottimizzazione dei corticosteroidi: farmaci come il vamorolone e il givinostat segnano il passaggio verso terapie più tollerabili e mirate.
In sintesi, la distrofia muscolare di Duchenne rappresenta oggi una condizione cronica complessa che richiede un impegno scientifico e clinico instancabile. Sebbene la sfida della guarigione definitiva rimanga aperta, la sinergia tra ricerca molecolare e cure standardizzate sta riscrivendo il futuro di migliaia di giovani pazienti, offrendo prospettive di vita precedentemente inimmaginabili.
Fonti:
- Distrofia muscolare di Duchenne – Orphanet ↩︎
- Duchenne Muscular Dystrophy (DMD) – Diseases ↩︎
- A Duchenne Muscular Dystrophy (DMD) Overview for Healthcare Professionals ↩︎
- Genotype–Phenotype Correlations in Duchenne and Becker Muscular Dystrophy Patients from the Canadian Neuromuscular Disease Registry – PMC – PubMed Central ↩︎
- About Duchenne Muscular Dystrophy – PTC Therapeutics ↩︎
- Distrofia muscolare di Duchenne e Becker – Orphanet ↩︎
- DMD gene: MedlinePlus Genetics ↩︎
- Duchenne Muscular Dystrophy – StatPearls – NCBI Bookshelf ↩︎
- Dystrophinopathies: Duchenne + Becker muscular dystrophy – Neuromuscular Home Page ↩︎
- Types of Genetic Variants – Parent Project Muscular Dystrophy ↩︎
- Advances in Duchenne Muscular Dystrophy: Diagnostic Techniques … ↩︎
- Correzione precisa delle mutazioni per delezione dell’esone nella distrofia muscolare di Duchenne mediante base e prime editing ↩︎
- Duchenne muscular dystrophy – Genes and Disease – NCBI – NIH ↩︎
- Muscular Dystrophy – StatPearls – NCBI Bookshelf – NIH ↩︎
- Distrofia Muscolare di Duchenne: cosa è, cause e cure | BGenetica ↩︎
- Distrofia muscolare di Duchenne e distrofia muscolare di Becker – Pediatria – MSD Manuals ↩︎
- Distrofia di Duchenne – Ospedale Pediatrico Bambino Gesù ↩︎
- Congenital Muscular Dystrophy – StatPearls – NCBI Bookshelf – NIH ↩︎
- Dystrophinopathies – StatPearls – NCBI Bookshelf – NIH ↩︎
- Therapeutic options for Duchenne muscular dystrophy: hope or … ↩︎
- Elevidys | European Medicines Agency (EMA) ↩︎
- EMA Rejects Marketing Authorization for Sarepta’s DMD AAV Gene Therapy ↩︎
- EMA Issues Negative Opinion for Sarepta’s Gene Therapy Elevidys – NeurologyLive ↩︎
- New treatment against Duchenne muscular dystrophy | European … ↩︎
- Is Givinostat (Duvyzat) Approved by EMA? – DMD Warrior ↩︎
- Parent Project aps ↩︎
- Distrofia muscolare di Duchenne: farmaci, terapie, sperimentazioni e qualità della vita ↩︎