La displasia fibrosa è un tumore osseo benigno, raro e di natura congenita, descritto per la prima volta da Von Recklinghausen nel 1891. La sua caratteristica fondamentale è la sostituzione del normale tessuto osseo con tessuto connettivo fibroso, anche noto come tessuto cicatriziale. Questa alterazione strutturale indebolisce l’osso interessato, rendendolo più suscettibile a deformità e fratture.
Caratteristiche
Per inquadrare correttamente la displasia fibrosa, è essenziale distinguerla da una neoplasia maligna. In medicina, un tumore, o neoplasia, è una massa di cellule che crescono e si dividono in modo incontrollato. La distinzione tra benigno e maligno si basa sul loro comportamento biologico:
- tumore benigno: la crescita della massa cellulare è circoscritta e non infiltrativa, ovvero non invade i tessuti circostanti. Fondamentalmente, è privo di capacità metastatizzante, cioè non si diffonde ad altre parti dell’organismo. La displasia fibrosa rientra in questa categoria;
- tumore maligno (cancro): è caratterizzato da una crescita rapida e infiltrativa. Le sue cellule hanno la capacità di staccarsi dalla massa originaria e diffondersi ad altri distretti dell’organismo attraverso il sangue o il sistema linfatico, dando origine a metastasi.
Il tessuto osseo sano è una struttura complessa e dinamica, progettata per fornire supporto meccanico, protezione e riserva di minerali. Al contrario, il tessuto fibroso che lo sostituisce nella displasia fibrosa è un ammasso disorganizzato di fibre collagene, privo di una funzione strutturale specifica. Questo tessuto è il risultato di un’alterazione a livello dei precursori delle cellule ossee. La mutazione genetica alla base della patologia impedisce la corretta maturazione dei preosteoblasti in osteoblasti funzionali; questi precursori, invece di produrre osso maturo, proliferano come cellule fibroblastiche disorganizzate. L’infiltrazione di questo tessuto anomalo altera l’architettura normale dell’osso, compromettendone l’integrità e la resistenza.

Eziopatogenesi
La displasia fibrosa è una malattia genetica congenita, cioè presente fin dalla nascita, ma è fondamentale sottolineare che non è ereditaria. Ciò significa che non viene trasmessa dai genitori ai figli, ma origina da un evento genetico che si verifica spontaneamente nell’individuo.

La causa della displasia fibrosa è una mutazione genetica post-zigotica, che avviene dopo il concepimento, durante i primi stadi dello sviluppo fetale. La mutazione interessa il gene GNAS1. Questo gene codifica per la subunità alfa della proteina Gs, un componente chiave nella trasduzione del segnale cellulare che regola la proliferazione e la differenziazione delle cellule ossee.
L’alterazione causata dalla mutazione porta a una stimolazione persistente dell’adenilciclasi e a un’alterata produzione dell’AMP ciclico. Questo segnale anomalo e costitutivamente attivo impedisce la corretta maturazione dei preosteoblasti, che invece di differenziarsi in osteoblasti sani e produrre tessuto osseo maturo, proliferano in modo disorganizzato formando il tessuto fibroso tipico della lesione.
Epidemiologia
La displasia fibrosa è una patologia rara che interessa principalmente bambini, adolescenti e giovani adulti. Le statistiche indicano che quasi il 75% dei pazienti ha meno di 30 anni, con un’età tipica di insorgenza compresa tra i 3 e i 15 anni. La prevalenza stimata nella popolazione generale è molto bassa, variando da 1 caso su 100.000 a 1 caso su 1.000.000 di individui.
Questa base eziopatogenetica comune si traduce in un’ampia varietà di manifestazioni cliniche, che definiscono i diversi quadri della malattia.

Clinica e localizzazioni comuni
La presentazione clinica della displasia fibrosa è estremamente variabile, spaziando da forme del tutto asintomatiche, scoperte casualmente, a quadri patologici complessi e disabilitanti. Le forme lievi possono rimanere silenti per tutta la vita, mentre quelle più severe sono associate a una sintomatologia significativa.
La displasia fibrosa viene classificata in due forme principali a seconda del numero di segmenti ossei coinvolti.
| Forma | Caratteristiche principali |
|---|---|
| Monostotica | È la forma più diffusa e interessa un solo osso. Le sedi più frequenti sono il femore, la tibia, le coste e le ossa mascellari. |
| Poliostotica | Colpisce più ossa e rappresenta una forma clinicamente più grave. Le ossa coinvolte possono appartenere allo stesso arto o a distretti diversi. |
In generale, le sedi scheletriche più comunemente colpite dalla displasia fibrosa includono: ossa del cranio e facciali, femore, tibia, coste, omero e bacino. Nelle forme monostotiche, le ossa mascellari sono i siti maggiormente coinvolti.
Nelle forme clinicamente significative, la displasia fibrosa può manifestarsi con un corteo sintomatologico caratteristico:
- dolore osseo: spesso è il sintomo principale, che può peggiorare con l’attività fisica;
- deformità ossee e asimmetrie: l’espansione del tessuto fibroso può causare rigonfiamenti e alterazioni della forma dell’osso, particolarmente evidenti a livello facciale o degli arti;
- aumentata tendenza alle fratture patologiche: l’osso indebolito può fratturarsi anche in seguito a traumi minimi;
- episodi di compressione nervosa: nelle localizzazioni cranio-facciali, l’espansione ossea può comprimere i nervi cranici, causando parestesie, disturbi visivi (calo o perdita della vista), uditivi e obliterazione dei seni nasali;
- scoliosi e dismetria degli arti: il coinvolgimento vertebrale o una crescita asimmetrica degli arti inferiori può portare a deviazioni della colonna e a differenze di lunghezza tra le gambe;
- manifestazioni cranio-facciali specifiche: possono includere occhi sporgenti (bulging eyes), malocclusione o disallineamento mascellare e denti disallineati o malformati.

Sindromi associate
Sebbene la displasia fibrosa si presenti spesso come una condizione isolata, può far parte di sindromi più complesse, dove la mutazione di GNAS1 coinvolge anche tessuti extra-scheletrici.

- Sindrome di McCune-Albright: rappresenta la forma più nota e complessa. È caratterizzata dalla triade classica di displasia fibrosa poliostotica, macchie cutanee caffè-latte e iperfunzione endocrina. Le manifestazioni endocrine più rilevanti includono:
- pubertà precoce (la più frequente);
- ipertiroidismo;
- eccesso di ormone della crescita (GH) con possibili quadri di acromegalia;
- ipercortisolismo neonatale (Sindrome di Cushing).
- Sindrome di Mazabraud: è una rara associazione tra displasia fibrosa (sia monostotica che poliostotica) e la presenza di mixomi intramuscolari, ovvero tumori benigni dei tessuti molli.
La valutazione di questi quadri clinici eterogenei richiede un percorso diagnostico strutturato per confermare la patologia e definirne l’estensione.
Percorso diagnostico
Una diagnosi accurata di displasia fibrosa richiede un approccio multidisciplinare che integri la valutazione clinica, la diagnostica per immagini e, in casi selezionati, esami istologici e genetici.
Il ruolo della diagnostica per immagini è cruciale e spesso sufficiente per porre il sospetto diagnostico.
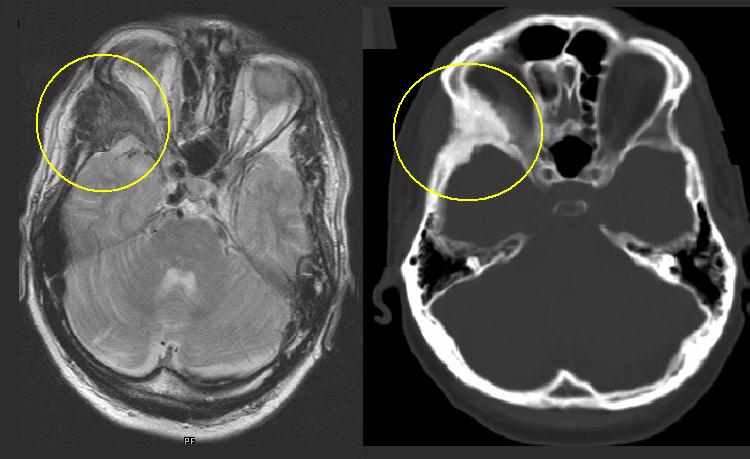
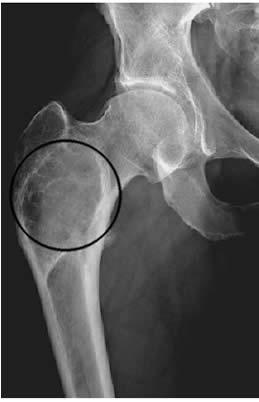
- Radiografia (RX): è quasi sempre il primo esame eseguito. L’aspetto radiografico tipico delle lesioni è quello a “vetro smerigliato” (ground glass), dovuto alla sovrapposizione di numerose trabecole ossee immature. I margini della lesione sono spesso sfumati e si fondono con l’osso sano circostante. L’aspetto può tuttavia variare, presentandosi anche come radiotrasparente (simil-cistico), sclerotico o misto.
- Tomografia Computerizzata (TC), in particolare Cone Beam (CBCT): questa metodica è superiore alla radiografia per la valutazione di regioni anatomiche complesse come il distretto cranio-facciale o il bacino. Permette una stima precisa dell’estensione della lesione e dei suoi rapporti con le strutture neurovascolari adiacenti.
- Risonanza Magnetica Nucleare (RMN): utile per la diagnosi differenziale con altre lesioni cistiche e per valutare le regioni ossee interessate.
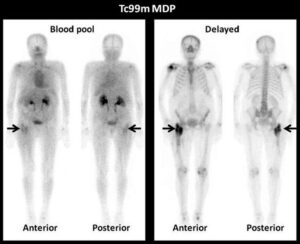
- Scintigrafia Ossea: è un esame funzionale fondamentale per determinare la distribuzione delle lesioni a livello dell’intero scheletro. Le aree affette da displasia fibrosa mostrano una marcata ipercaptazione del radiofarmaco (definite “zone calde”), rendendo questo esame indispensabile per distinguere la forma monostotica da quella poliostotica.

La biopsia ossea non è sempre necessaria, ma diventa fondamentale nei casi dubbi, per la diagnosi differenziale (es. con osteosarcoma a basso grado) o in caso di sospetta trasformazione maligna. A causa del mosaicismo somatico della mutazione, l’analisi del sangue periferico può produrre falsi negativi. Pertanto, il gold standard per la conferma genetica della mutazione di GNAS1, specialmente nei casi monostotici o diagnosticamente complessi, è l’analisi del tessuto affetto ottenuto tramite biopsia.
I test di laboratorio possono supportare la diagnosi e il monitoraggio. È comune riscontrare un aumento delle fosfatasi alcaline, indice di un elevato turnover osseo. Nei pazienti con forme poliostotiche o sindromiche, è cruciale valutare il metabolismo calcio-fosforo e l’assetto ormonale completo. Un reperto chiave è la possibile ipofosfatemia da ipersecrezione di FGF23. Questa molecola è sovraprodotta dalle cellule staminali scheletriche mutate e causa una perdita renale di fosfati. Sebbene meccanismi compensatori rendano rara l’ipofosfatemia franca, essa rappresenta un fattore critico nello sviluppo di rachitismo/osteomalacia in pazienti con un elevato carico di malattia scheletrica.

Gestione terapeutica
Essendo una malattia genetica a mosaico, non esiste una cura risolutiva per la displasia fibrosa. L’obiettivo del trattamento è quindi sintomatico e mira a ridurre il dolore, prevenire le fratture, correggere le deformità e gestire le complicanze.
L’approccio terapeutico varia in base alla gravità e alla sintomatologia.
- Forme lievi/asintomatiche: generalmente non richiedono un trattamento attivo. È sufficiente un monitoraggio periodico (osservazione o watchful waiting) con esami radiografici.
- Forme gravi/sintomatiche: richiedono un intervento attivo, che può essere di tipo farmacologico, chirurgico o una combinazione di entrambi.
La gestione della displasia fibrosa richiede un approccio personalizzato e multidisciplinare.
| Approccio terapeutico | Obiettivo primario | Esempi e considerazioni chiave |
|---|---|---|
| Terapia farmacologica | Riduzione del dolore, rinforzo osseo, inibizione del riassorbimento osseo. | Bifosfonati (es. Pamidronato, Acido Zoledronico): riducono il dolore e il turnover osseo inibendo l’attività degli osteoclasti. Denosumab: alternativa che inibisce l’attività osteoclastica. Può essere efficace, ma la sua sospensione è associata a un rischio di effetto rebound e ipercalcemia. |
| Terapia chirurgica | Correzione di deformità, stabilizzazione di fratture, decompressione nervosa, miglioramento estetico. | Approcci conservativi (curettage, rimodellamento) presentano un rischio di recidiva. L’escissione radicale con ricostruzione offre maggiori tassi di successo. La strategia dipende dalla localizzazione, come nella classificazione di Chen e Noordhoff per il distretto cranio-facciale: Zona 1 (fronto-orbitale) consente resezioni radicali; Zona 2 (regioni craniche coperte) consente resezioni radicali, ma in grado minore rispetto alla zona 1 Zona 3 (base cranica centrale) richiede approcci conservativi per il rischio a nervi e vasi; Zona 4 (area dentata) predilige metodi conservativi per preservare la funzione dentale. |
| Supporto e riabilitazione | Mantenimento della funzione, prevenzione delle fratture, gestione della qualità di vita. | Uso di tutori protettivi, fisioterapia (inclusa idrochinesiterapia) per ottimizzare forza e mobilità, adeguato apporto di calcio e vitamina D, e supporto psicologico. |
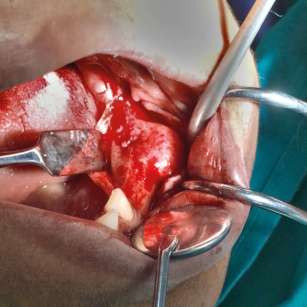
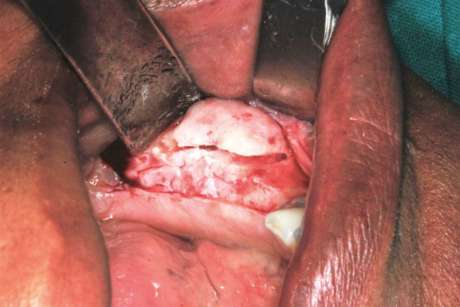

Prognosi e complicanze
La prognosi della displasia fibrosa dipende direttamente dalla gravità e dall’estensione della malattia. In generale, la patologia tende a stabilizzarsi con il raggiungimento della maturità scheletrica.
Per le forme lievi e monostotiche, la prognosi è eccellente, consentendo una qualità di vita del tutto normale. Al contrario, per le forme poliostotiche gravi o sindromiche, la prognosi è più complessa. Le complicanze possono essere significative, impedendo ai pazienti di condurre una vita normale e richiedendo un monitoraggio e un trattamento continui.
Caricamento….
Le forme gravi possono portare a diverse complicanze a lungo termine:
- deformità gravi e fratture ricorrenti: derivano dalla debolezza strutturale dell’osso fibroso e possono limitare gravemente la mobilità;
- deficit neurologici: nelle forme cranio-facciali, la compressione dei nervi cranici (es. ottico, acustico) può causare un calo progressivo o la perdita irreversibile della vista e dell’udito;
- artrite: quando le lesioni si trovano in prossimità di un’articolazione, le deformità ossee che ne derivano possono alterare la normale meccanica articolare, portando a un’artrite secondaria;
- trasformazione maligna: si tratta di un’eventualità molto rara, ma temibile. Sebbene stime più datate riportassero cifre più elevate, il consenso attuale si attesta su una frequenza inferiore all’1%. Il tumore più comune che ne può derivare è l’osteosarcoma. Segni clinici che devono imporre un’indagine urgente includono un rapido incremento delle dimensioni della lesione, l’insorgenza di dolore in una lesione precedentemente asintomatica, o una netta intensificazione di un dolore preesistente.
Una comprensione approfondita di questi aspetti è fondamentale per la gestione clinica del paziente.
Fonti: