Una tecnica molto utilizzata nei laboratori è l’elettroforesi che si basa sulla capacità di movimento di molecole più o meno grandi quando vengono sottoposte a un campo elettrico. Due esempi importanti di macromolecole che possono essere separati tra di loro sfruttando questo principio sono gli acidi nucleici e le proteine. Proprio queste due macromolecole perché in determinate condizioni di pH, che può essere anche a pH fisiologico, si possono avere molecole che presentano carica netta diversa da zero e quindi possiamo utilizzare un sistema in cui applichiamo una differenza di potenziale, un campo elettrico, per poterle separare. Le sostanze che possono essere separate con questa tecnica sono:

- amminoacidi singoli;
- peptidi;
- proteine;
- nucleotidi;
- DNA, RNA;
Nell’elettroforesi in fase libera (o frontale) le particelle cariche si muovono attraverso una soluzione (poco attrito, elevata velocità di migrazione). Nell’elettroforesi su supporto (o zonale) le particelle si muovono attraverso un mezzo poroso che può essere agarosio, poliacrilamide oppure carta. La tecnica separativa elettroforetica che maggiormente viene utilizzata in tutti i laboratori del mondo è l’elettroforesi su supporto che permette un maggior grado di risoluzione e separazione delle nostre macromolecole. Si tratta di una tecnica essenzialmente di tipo analitico ma anche preparativo.
Principi generali:
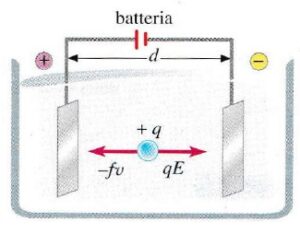
- Una molecola con carica netta non nulla posta tra due elettrodi di segno opposto migra verso l’elettrodo con segno opposto alla sua carica netta;
- La molecola si muove verso l’elettrodo di segno opposto con una velocità che è proporzionale all’entità del campo elettrico e della sua carica ed inversamente proporzionale all’attrito che incontra nel muoversi.
Now loading…
Elettroforesi zonale
L’elettroforesi zonale permette la separazione dei componenti di una miscela in zone o bande poiché per effetto del supporto solido, altri fattori come l’effetto setaccio, esaltano la differenza di mobilità anche tra particelle con densità di carica uguale. Si ha filtrazione molecolare, fenomeno dovuto al reticolo tridimensionale delle catene che costituiscono il gel: agar, amido o poliacrilamide. Le sostanze ad alto peso sono ostacolate nella migrazione più di quelle a peso molecolare più basso. Il campione deve essere depositato tutto in un’unica zona del supporto e a partire da quella zona avviene la migrazione.
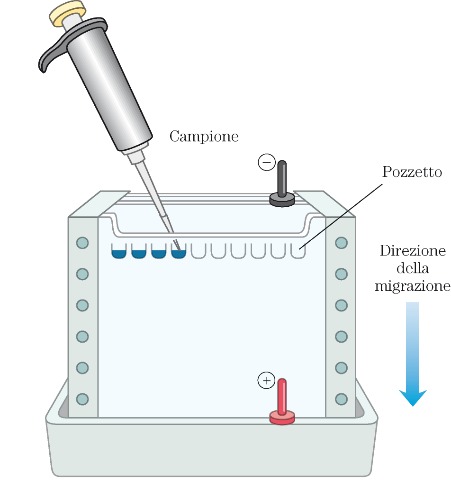
Elettroforesi verticale
Nella figura a destra è riportato uno schema tipico di apparato elettroforetico che è costituito da una cosiddetta cella elettroforetica in cui si posiziona il supporto. Generalmente per l’elettroforesi verticale si utilizza il gel di poliacrilammide come supporto. La poliacrilamide è una sostanza che polimerizza, assume la consistenza di gel, e questo gel viene mantenuto tra due supporti rigidi che sono generalmente due vetrini che devono essere trasparenti per seguire la corsa elettroforetica. Sul fronte del gel vengono creati degli alloggiamenti chiamati pozzetti in cui si depositano, tramite una micropipetta, i campioni. La parte essenziale della strumentazione elettroforetica è il generatore di energia elettrica attraverso il quale viene applicata la differenza di potenziale che crea il campo elettrico che permette poi la migrazione di queste molecole cariche. Quindi ci sarà un polo negativo e un polo positivo, rispettivamente catodo e anodo e si assisterà alla migrazione verso uno dei due poli a seconda della carica iniziale della nostra molecola.
Elettroforesi orizzontale
Caricamento…
Un altro tipo di tecnica elettroforetica di tipo zonale è l’elettroforesi orizzontale. Il gel utilizzato per questo tipo di elettroforesi che permette la separazione degli acidi nucleici è fatto di agarosio, il cui grado di risoluzione è inferiore rispetto a quello della poliacrilamide. Si presta meglio per gli acidi nucleici perché le dimensioni molecolari di un acido nucleico rispetto a una proteina sono, nella maggior parte dei casi, maggiori. Il principio è sempre lo stesso: vi è la celletta elettroforetica in cui si allestisce il campione, si mette il gel che questa volta è posizionato orizzontalmente con dei pozzetti che rappresentano il fronte del gel, cioè la parte del gel da cui comincia la corsa. In tutte le celle elettroforetiche i due elettrodi, rispettivamente negativo e positivo (catodo e anodo), sono sempre presenti con due determinati colori: il rosso rappresenta il positivo, il nero è il negativo. Quindi quando viene generata la differenza di potenziale si genera il campo elettrico e si avrà la migrazione del campione. Altri esempi di supporti solidi possono essere carta, acetato di cellulosa oppure i gel; questi ultimi sono quelli più utilizzati e possono essere di amido, agar, agarosio o poliacrilamide. Quelli più utilizzati sono agarosio e poliacrilamide. L’agarosio purificato è molto usato per la separazione di proteine ad alto peso molecolare e di acidi nucleici mentre la poliacrilamide è un polimero sintetico dovuto alla reazione di polimerizzazione tra acrilamide e bisacrilamide in presenza di catalizzatore e iniziatore.

Gel di agarosio
Caricamento…
Per preparare il gel di agarosio si usa la polvere di agarosio con una preparazione abbastanza semplice rispetto a quella della poliacrilammide. È una reazione di solidificazione dell’agarosio mentre, invece, diversa è la preparazione della poliacrilamide dove abbiamo una reazione di polimerizzazione delle molecole di acrilamide mescolate con la bisacrilamide, un derivato dell’acrilamide che serve per formare dei legami incrociati tra le varie maglie del gel. Per velocizzare la reazione di gelificazione dell’agarosio si espone il gel a temperature più basse, nel frigorifero a 4°C. All’atto pratico per fare un gel di agarosio commercialmente sono disponibili tanti tipi di polvere di agarosio che differiscono tra di loro per il grado di purezza.
Il gel si prepara pesando una certa quantità di agarosio in polvere che poi si scioglie in un tampone indicato generalmente con la sigla TBE o TAE che sta per tris-borato-EDTA o tris-acetato-EDTA e sono questi i tamponi che vengono utilizzati per solubilizzare questa polvere di agarosio in una beuta o in un becher. La soluzione viene fatta bollire posizionando su delle piastre riscaldanti con agitatore magnetico per poter disciogliere le soluzioni. Una volta che diventa liquido si aggiunge un agente tracciante che ci permette di visualizzare la separazione delle varie bande che corrispondono ciascuna a un segmento dell’acido nucleico che stiamo andando ad analizzare. In seguito si versa il gel nell’apposito apparato per la corsa elettroforetica.
Per gli acidi nucleici l’agente tracciate più utilizzato, anche se attualmente non viene più utilizzato perché molto tossico, è l’etidio bromuro, un colorante fluorescente intercalante che consente di visualizzare il DNA. Ha un picco di assorbimento a 250 nm ed emette nel visibile, riduce la velocità di migrazione del DNA del 15%. È molto tossico e altamente cancerogeno in quanto per la modalità con cui interagisce con gli acidi nucleici è un agente intercalante, cioè si posiziona al centro della doppia elica del DNA, ed è eccitabile tramite raggi ultravioletti quindi poi si posiziona il gel su un transilluminatore con lampada ad UV, viene eccitato dagli UV e questo apparecchio permette di visualizzare le bande in maniera fluorescente. Dato che l’etidio bromuro è un agente intercalante è molto pericoloso perché appunto si intercala e quindi è mutageno. Proprio per questa pericolosità dell’etidio bromuro sono stati introdotti commercialmente altri agenti intercalanti che si intercalano soltanto con l’acido nucleico che è stato purificato e quindi sono meno dannosi.
Dopo aver aggiunto l’etidio bromuro il gel che è liquido si cola all’interno della cella elettroforetica in una vaschetta che ha dei margini in maniera tale che non coli da una parte e dall’altra e poi si attende che si solidifichi e il gel è pronto. Per creare i pozzetti si aggiunge un pettinino in maniera tale da formare i pozzetti.

In seguito all’applicazione di un campo elettrico, le molecole di DNA che sono cariche negativamente (perché sono degli acidi) migreranno dall’elettrodo negativo verso il positivo quindi verso l’anodo. I frammenti più piccoli migreranno molto più rapidamente quindi i vari segmenti di acido nucleico vengono separati in base alla dimensione molecolare: quelli più piccoli li ritroveremo più in basso mentre quelli più grandi si fermeranno prima. In base alla dimensione molecolare degli acidi nucleici che noi dobbiamo andare a separare utilizziamo delle differenti concentrazioni di agarosio perché maggiore è la concentrazione di agarosio, minore sarà la grandezza dei pori che si formeranno all’interno dell’agarosio. Più è fitta questa rete, più è ostacolata la migrazione dell’acido nucleico mentre, invece, se abbiamo degli acidi nucleici con peso molecolare molto alto, dobbiamo utilizzare una concentrazione di agarosio molto bassa.
La velocità di migrazione è influenzata da: dimensione del DNA, concentrazione di agarosio nel gel, conformazione del DNA, voltaggio applicato, presenza di etidio bromuro, composizione ionica del buffer.
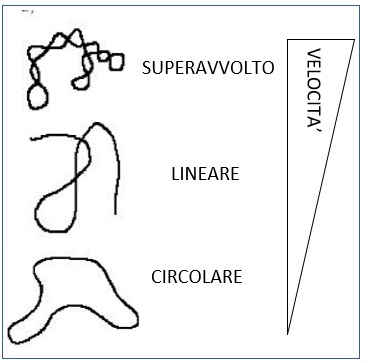
Il DNA può trovarsi in diverse conformazioni che hanno differenti velocità di migrazione anche se di dimensione uguale:
- la forma superavvolta corre più velocemente in quanto più compatta.
- La forma circolare corre più lenta perché “più ingombrante” e fa più fatica a muoversi all’interno dei pori del gel
- La forma lineare ha una mobilità intermedia (la forma lineare è, per esempio, quella che si ritrova come prodotto nella PCR)
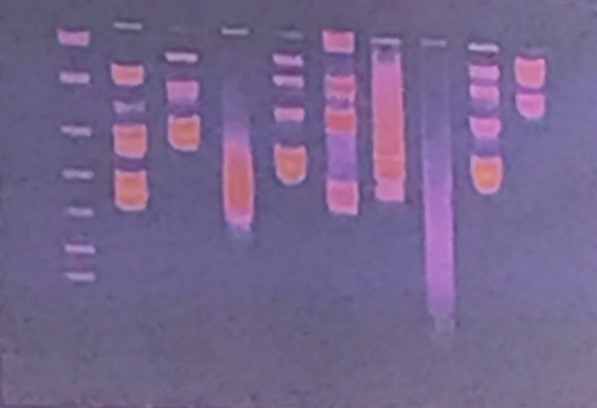
Questa è un’immagine tipica che si ottiene su transilluminatore, cioè quando il gel viene posizionato su questo strumento che ha una lampada a UV sul basso. Questo è il segnale dovuto all’etidio bromuro che si intercala tra gli acidi nucleici. Generalmente nel primo pozzetto (il primo a sinistra nella figura) viene caricato un marker di peso molecolare. Se noi vogliamo stabilire il peso molecolare del nostro segmento di acido nucleico dobbiamo paragonarlo a un marker di peso molecolare che è una miscela di frammenti di DNA con un peso molecolare noto e quindi dall’altezza di migrazione del nostro campione possiamo risalire al peso molecolare e quindi valutare la qualità del nostro campione iniziale.

Gel di poliacrilammide
L’elettroforesi su gel di poliacrilammide è utilizzata maggiormente nella separazione delle proteine perché presenta un grado di risoluzione maggiore rispetto all’agarosio essendo le proteine più piccole rispetto agli acidi nucleici. Però l’agarosio può essere utilizzato anche per le proteine e la poliacrilamide per gli acidi nucleici in quanto non c’è questa separazione nettissima.
Alla fine nell’elettroforesi su gel di poliacrilammide ogni banda nera (in molti casi è blu perché il colorante maggiormente utilizzato per la colorazione delle proteine è il blue di coomassie) corrisponderà a una differente proteina.
Il gel di poliacrilamide funziona come un setaccio molecolare: si formano delle maglie all’interno all’acrilamide in seguito a una reazione di polimerizzazione. Applichiamo sul pozzetto, cioè sul fronte del gel, la nostra miscela e la velocità di migrazione è funzione dell’inverso della dimensione molecolare, cioè quelle più piccole migreranno più velocemente perché le maglie del gel si lasciano attraversare più facilmente, mentre quelle più grandi incontrano un ostacolo maggiore alla migrazione. Quindi le proteine che si trovano più in alto sono quelle con una dimensione molecolare maggiore e via via decrescono verso il basso.
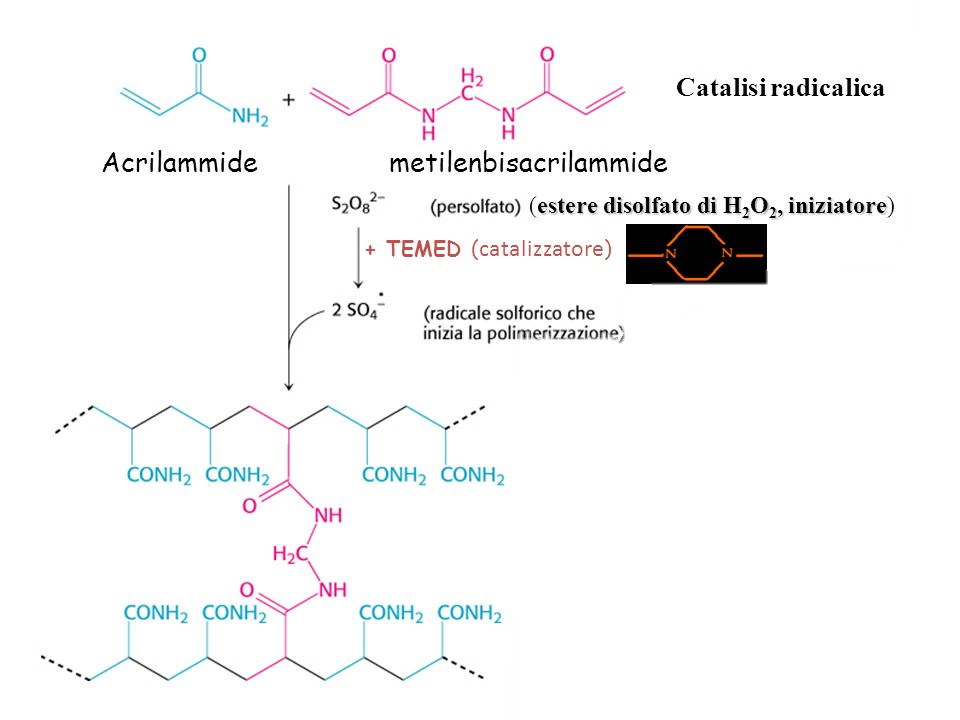
Nella figura affianco viene illustrata la reazione di polimerizzazione. Per poter preparare un gel di poliacrilamide si miscelano varie sostanze tra di loro, tra cui le principali sono l’acrilamide che si miscela con metilenbisacrilamide. Un tempo l’acrilamide e la metilen bisacrilamide si preparavano separatamente, adesso sono disponibili commercialmente le miscele liquide di acrilamide e di metilen bisacrilamide già pronte con una percentuale differente e la cui proporzione varia a seconda della porosità che noi vogliamo ottenere nel gel. L’acrilamide in polvere è neurotossica per il nostro organismo, e quindi bisogna dotarsi di mascherina, occhiali, guanti. Ora è disponibile l’acrilamide in soluzione che è meno pericolosa della polvere la quale si può inalare e può andare a contatto con la pelle. La reazione di polimerizzazione tra l’acrilamide e la metilen bisacrilamide ha bisogno di un input con un iniziatore radicalico perché si tratta di una polimerizzazione radicalica e un catalizzatore rappresentato dal TEMED. L’iniziatore radicalico è il persolfato che commercialmente è disponibile come ammonio persolfato. Aggiungendo nel nostro becher queste due sostanze, cioè TEMED e ammonio persolfato, diamo il via alla reazione di polimerizzazione. Quando noi stiamo facendo il gel e aggiungiamo queste due sostanze dobbiamo essere molto veloci nel colare il gel, cioè nel posizionare e versare il gel attraverso i due vetrini che poi contengono il gel altrimenti la polimerizzazione avviene nel becher. La percentuale di acrilamide può variare tra il 20%, 15%, 5% e 3,5%. La percentuale di acrilamide più bassa permette di separare delle proteine più grandi e viceversa.
Caricamento….
SDS-PAGE
Un tipo particolare di tecnica elettroforetica è l’SDS-PAGE, universalmente conosciuta e utilizzata in tutti i laboratori del mondo che studiano proteine. Questa particolare tecnica elettroforetica di studio delle proteine si basa sull’utilizzo di un blando detergente anionico chiamato SDS (sodio dodecil solfato). Esso ha delle cariche negative e legandosi alle proteine da analizzare, le carica tutte allo stesso modo, cioè vengono uniformate dal punto di vista della carica. Il principio di separazione delle proteine diventa soltanto il peso molecolare: le proteine che si fermano più in alto nel gel avranno peso molecolare maggiore rispetto a quelle che migrano più in basso nel gel. E’ importante che le molecole abbiano tutte esattamente lo stesso rapporto carica/massa (migrazione in base al solo peso molecolare). Ciò è normale per i frammenti di acidi nucleici in cui il numero di cariche negative dovute ai gruppi fosfato è esattamente proporzionale alla lunghezza. Per gli acidi nucleici si usano gel al 4%-6% per la separazione di frammenti molto piccoli. In realtà questo tipo di tecnica è molto utilizzata nella separazione delle proteine ed è un tipo di tecnica elettroforetica di tipo denaturante. PAGE sta per PoliAcrilamide Gel Elettroforesi.
Elettroforesi denaturante significa che la proteina viene analizzata nel suo stato denaturato, mantiene soltanto la propria dimensione molecolare. Da questo tipo di tecnica non otteniamo nessun tipo di informazione riguardante la struttura della proteina, cioè la andiamo a studiare solo per quanto riguarda il peso e in base al peso si risale al tipo di proteina. Se la proteina è conosciuta, e attraverso altri metodi di analisi andiamo a determinarne la struttura.

Prima di poter caricare il campione sul gel, le proteine vanno incontro a un processo di denaturazione. Vengono utilizzati dei reagenti denaturanti. L’SDS in sé conferisce a tutte le proteine la carica negativa e vengono anche parzialmente denaturate, però poi nella nostra soluzione vanno aggiunti altri agenti denaturanti di cui i maggiormente utilizzati sono l’urea che ad alte concentrazioni dissocia le subunità, il mercaptoetanolo la cui sigla è β-SH e il DTT che è il ditiotreitolo. La specificità di queste sostanze è la presenza dello zolfo sotto forma di gruppi solfidrilici che va a rompere i ponti di disolfuro che tengono insieme le varie subunità polipeptidiche nel caso in cui ci siano questi tipi di interazioni covalenti. Questo perciò è il primo passaggio da fare per poter separare le proteine tramite tecnica elettroforetica. L’SDS denatura completamente le proteine inserendosi con la porzione apolare verso l’interno e con la porzione carica negativamente all’esterno. Le proteine così risultano avvolte in un guscio di cariche negative uniformi. Poiché la quantità di SDS usato è in rapporto di 1:2, cioè una molecola di SDS ogni due amminoacidi, la mobilità della stessa dipenderà dalla lunghezza della catena polipeptidica e quindi dal peso molecolare mentre il rapporto carica/massa sarà uguale per tutte.
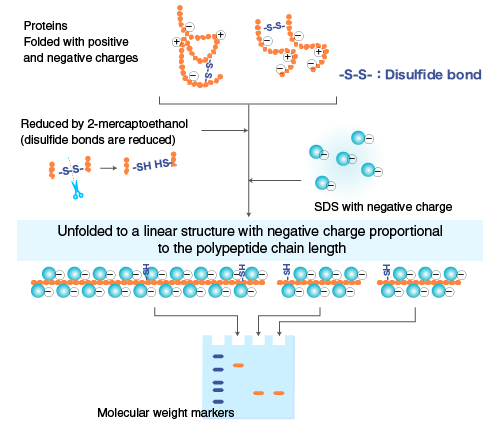
Nell’immagine sono illustrati i vari passaggi. A sinistra vengono rotti i ponti di disolfuro tra le proteine con gli agenti denaturanti e si aggiunge SDS ottenendo una condizione di questo tipo per la proteina: tutte le proteine sono caricate negativamente dalla presenza dell’SDS e vengono così caricate sul gel dove ogni banda corrisponde a una singola proteina. Abbiamo la corsa elettroforetica, cioè l’allestimento della cella elettroforetica, l’aggiunta del tampone di corsa o buffer di corsa. Generalmente il più utilizzato è il buffer di corsa costituito da tris-HCl e Glicina. Poi alla fine si ha la visualizzazione delle bande proteiche sul nostro gel. Quindi il risultato finale tipico può essere quello dove le proteine al termine della corsa elettroforetica vengono colorate e la colorazione generalmente si fa con il coomassie brilliant blue g-250. Ci sono due tipi di blue coomassie: il blue coomassie g-250 e il blue coomassie r-250. La differenza è la presenza di alcuni gruppi aggiuntivi nella forma g che è quella più utilizzata ed è maggiormente risolutiva rispetto alla forma r. Questo colorante ha la capacità di reagire con alcuni particolari amminoacidi della catena polipeptidica e rimane attaccato alle proteine. Con questa tecnica noi utilizziamo come discriminante, cioè come elemento di differenziazione delle varie proteine, soltanto il peso molecolare perché tutte le proteine sono cariche allo stesso modo avendole prima trattate con l’SDS. Per poter poi risalire al peso molecolare della nostra proteina sconosciuta possiamo paragonare la distanza di migrazione in cm rispetto al fronte del gel che è sempre la porzione del gel più in alto perché l’SDS-PAGE è un tipo di tecnica che si fa con elettroforesi verticale quindi dall’alto verso il basso. Le proteine più in alto hanno un peso maggiore di quelle poste in basso. Si può essere molto più precisi nel determinare il peso molecolare perché esiste una relazione matematica molto precisa tra il logaritmo della massa relativa della proteina e la sua distanza in cm. È una correlazione inversa perché maggiore è la migrazione minore sarà il peso.
Man mano che procediamo nel processo di purificazione della proteina aumenta la percentuale della nostra proteina di interesse perché maggiormente presente rispetto alle altre.
Caricamento….
Applicazioni della SDS-PAGE:
- determinare la purezza del campione;
- determinare il peso molecolare del campione;
- presupposto per una successiva analisi, il western blotting o western blot che permette di analizzare nel dettaglio una singola proteina utilizzando anticorpi specifici contro quella determinata proteina;
- determinare la purificazione della proteina. Per esempio quando alla fine otteniamo il nostro gel con le nostre bande proteiche che sono colorate e che possiamo visualizzare con il blue coomassie, possiamo purificare quella proteina tagliando quella regione del gel che contiene la nostra proteina e utilizzandola per successive analisi fatte in spettrometria di massa, la tecnica analitica che meglio si presta per l’identificazione precisa di una proteina.
Metodi di colorazione delle proteine dopo la separazione elettroforetica:
- Coomassie Brilliant Blue (CBB);
- Silver staining – colorazione con ioni di argento. Questo tipo di colorazione rispetto alla colorazione con coomassie brilliant blue g ha un grado di sensibilità maggiore e riesce a rilevare delle quantità di proteine più piccole nel nostro gel;
- Colorazione di Schiff con acido periodico (PAS) – non viene utilizzata quasi mai nei laboratori di ricerca;
- Densitometria a scansione – il densitometro è uno strumento che permette di tradurre il nostro segnale che è la nostra banda in un numero. È una specie di scanner attraverso cui le varie bande sono attraversate da un fascio di luce e dall’analisi fatta con un densitometro viene fuori un profilo densitometrico: a ogni proteina corrisponde un picco che è più o meno elevato a seconda dell’intensità della banda. Quando si fanno delle analisi chimico-cliniche e quindi l’elettroforesi delle proteine sieriche se si legge il risultato vi è il profilo delle varie ammine o globuline dove l’albumina sierica è quella con il picco maggiore perché è quella più presente a livello del siero. Quindi un profilo di questo tipo viene fuori dall’analisi densitometrica.
Caricamento….
Elettroforesi NATIVA
L’elettroforesi nativa, come dice il termine, è una tecnica separativa delle proteine che però preserva la conformazione nativa della proteina. È utilizzata ogni qualvolta è importante conservare l’attività funzionale delle proteine (l’SDS-PAGE è un’elettroforesi in condizioni denaturanti). Un esempio è la separazione degli isoenzimi della lattico deidrogenasi LDH del siero umano. Esistono 5 isoenzimi le cui differenze nella struttura delle subunità produce una diversa carica elettrica (stesso peso molecolare). Questa differenza di carica consente la separazione dei 5 isoenzimi della LDH quando si esegue l’elettroforesi. Dopo la corsa elettroforetica gli isoenzimi presenti nel gel sono evidenziati per mezzo delle reazioni riportate affianco.
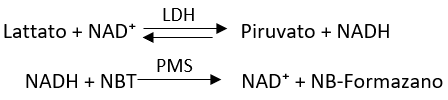
Nella prima reazione del lattato con il NAD⁺ tramite il lattato deidrogenasi (LDH) si forma il piruvato e il NADH. Il NADH che viene prodotto viene evidenziato con il composto NBT in presenza di PMS per formare la forma ossidata del NAD più NB-Formazano che è un prodotto che si colora di blu-marroncino quindi è una misura indiretta dell’attività dell’enzima, cioè un metodo colorimetrico.
Il risultato che viene fuori è riportato nel cartoon sottostante dove sono riportati a sinistra le 5 isoforme del LDH: la cui carica elettrica positiva aumenta andando da LDH1 a LDH5. Possiamo ottenere dalla densità delle bande il profilo elettroforetico.
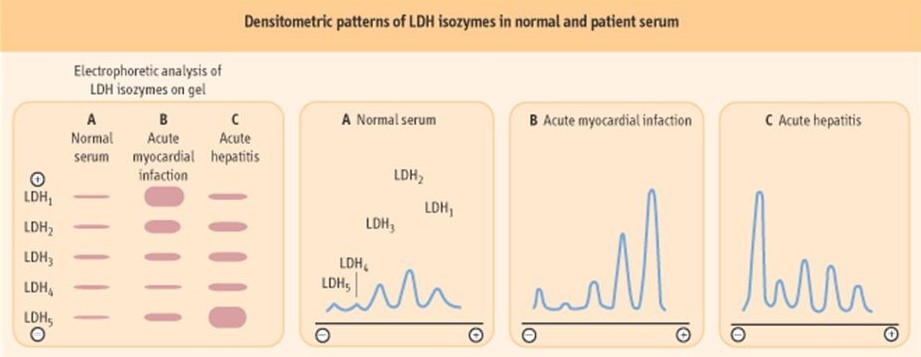

Sempre nella tabella, in A abbiamo un siero normale, in B un infarto acuto del miocardio dove aumenta notevolmente LDH1 mentre in C abbiamo un’epatite acuta dove aumenta LDH5 che è tipica dell’epatocita. Nei cardiomiociti si trova più che altro l’isoforma numero 1. LDH1 e LDH5 possono essere indice rispettivamente dell’infarto o dell’epatite acuta perché in questi casi si ha necrosi cellulare e quindi fuoriuscita del contenuto della cellula e si ritrovano questi isoenzimi a livello del siero dove normalmente non sono presenti, cioè vi è un aumento delle isoforme e in base al tipo di isoforma che aumenta si può valutare l’infarto acuto del miocardio oppure l’epatite acuta. Ci sono anche altri tipi di marker enzimatici che vengono utilizzati per valutare questi tipi di danni ma l’LDH rimane uno degli enzimi maggiormente utilizzati per queste analisi.
Caricamento….