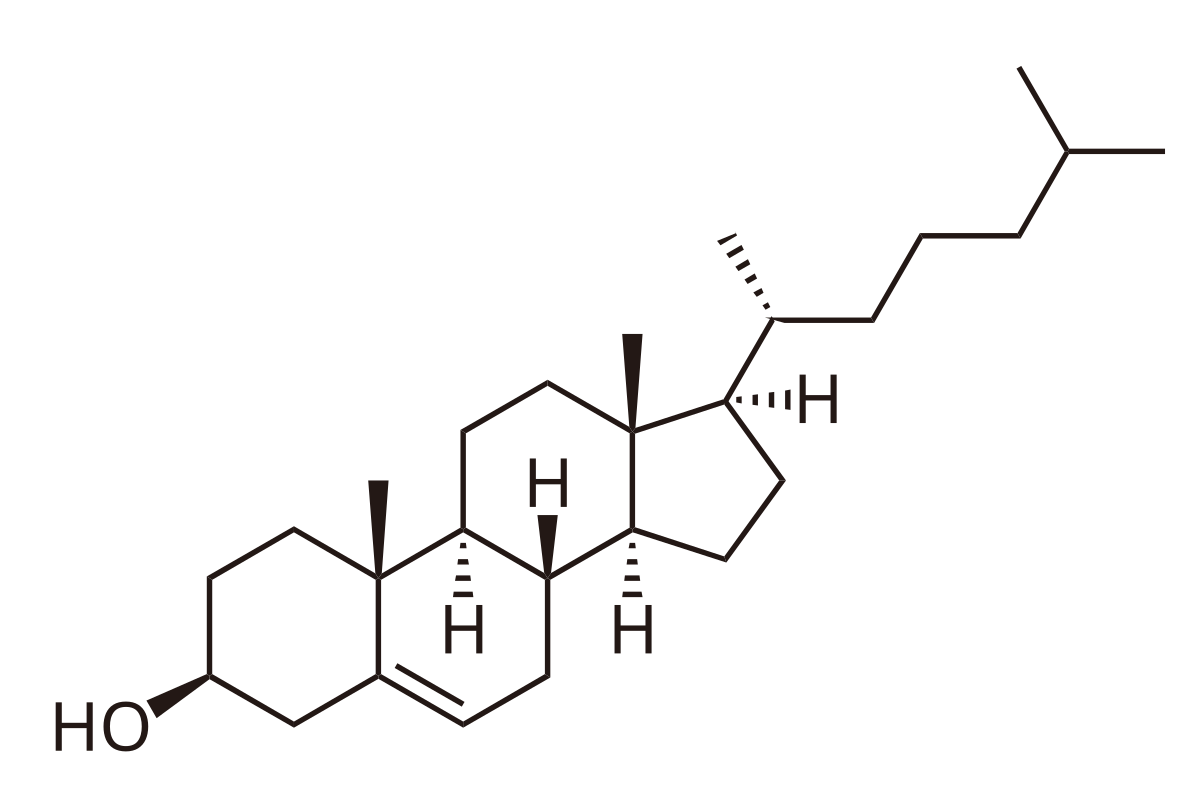
Il colesterolo è un lipide utilizzato come componente delle membrane cellulari (colma alcune lacune formate dai fosfolipidi per la presenza di acidi grassi insaturi), precursore degli ormoni steroidei (cortisolo, aldosterone, ormoni sessuali), di vitamina D e degli acidi biliari (ruolo detergente che aumenta la superficie di esposizione dei lipidi alimentari per poter essere attaccati dalle lipasi intestinali). Esso è presente negli alimenti di origine animale, tuttavia viene principalmente prodotto per via endogena. Possiede 27 atomi di C disposti a formare 4 anelli chiusi (A, B, C, D) fusi tra loro. Presenta inoltre un gruppo -OH sul C3, un doppio legame tra C5-C6, una catena idrocarburica a 8 atomi di carbonio su C17 e 2 gruppi metilici su C10 e C13. Tutti i carboni hanno tutti come unico precursore l’acetato. Durante la sintesi del colesterolo vengono prodotti degli intermedi isoprenoidi che sono precursori di altri lipidi.
Il colesterolo si forma in 4 tappe a partire dall’acetil-Coenzima A (Acetil-CoA):
Condensazione di tre unità di acetato per formare un intermedio a 6 atomi di carbonio (mevalonato);
Conversione del mevalonato in unità isopreniche;
Polimerizzazione di unità isopreniche a 5 atomi di carbonio per formare lo squalene, composto lineare a 30 atomi di carbonio;
Ciclizzazione dello squalene per formare il nucleo steroideo a quattro anelli, da cui, attraverso una serie di altre modifiche si forma il colesterolo.
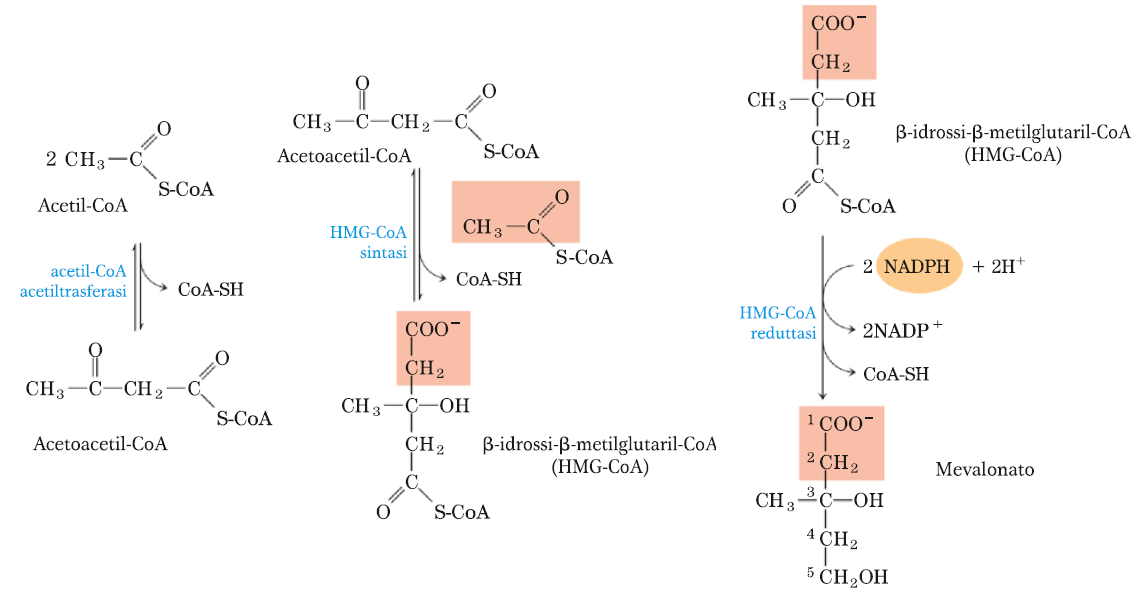
TAPPA 1 – SINTESI DEL MEVALONATO DALL’ACETATO
Due molecole di acetil-CoA condensano formando acetoacetil-CoA con liberazione di una molecola di Coenzima A ridotto. La molecola di acetoacetil-CoA prodotta reagirà con una terza molecola di acetil-CoA per generare il β-idrossi-β-metilglutaril-CoA (HMG-CoA) con liberazione di CoA-SH. Queste reazioni sono catalizzate rispettivamente dalla acetil-CoA acetiltrasferasi (una tiolasi) e dalla HMG-CoA sintasi. Infine l’enzima HMG-CoA reduttasi riduce l’HMG-CoA a mevalonato, il donatore di e– è il NADPH.
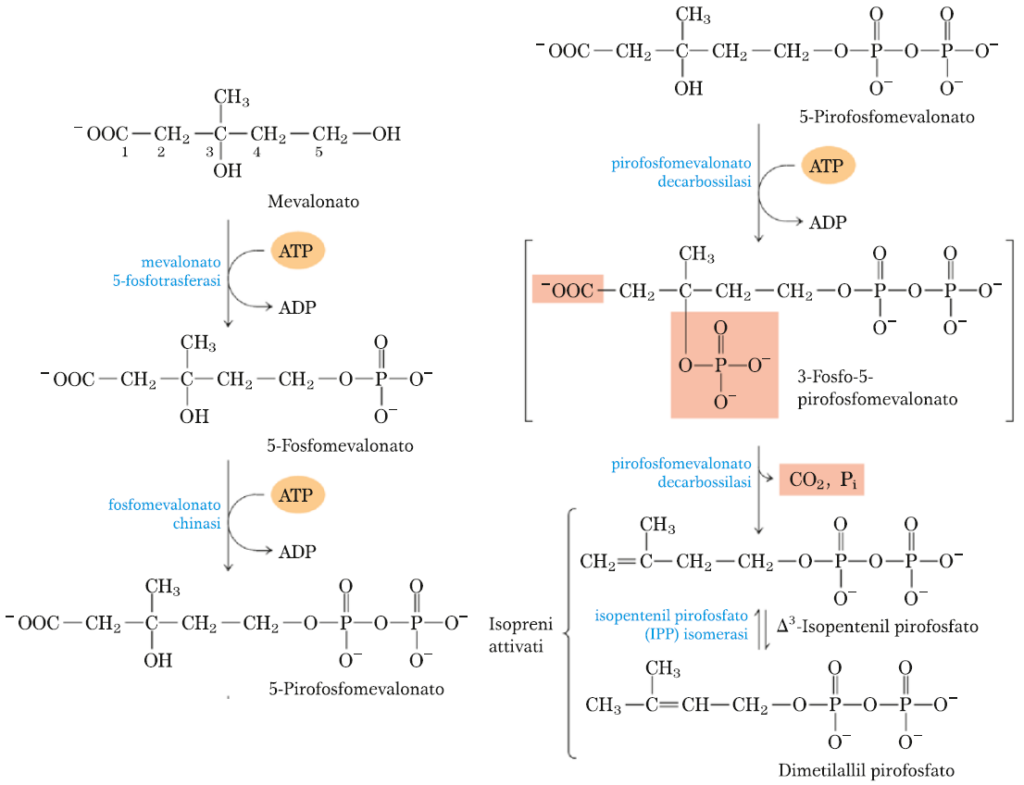
TAPPA 2 – CONVERSIONE DEL MEVALONATO IN DUE UNITA’ ISOPRENICHE ATTIVATE
3 gruppi fosfato vengono trasferiti da 3 molecole di ATP al mevalonato, formando l’intermedio 3-fosfo-5-pirofosfomevalonato. La prima reazione che avviene è l’aggiunta di un gruppo fosfato in posizione del carbonio 5 formando 5-fosfomevalonato tramite l’enzima mevalonato 5-fosfotrasferasi che utilizza una molecola di ATP e libera ADP. La seconda reazione è l’aggiunta di un nuovo gruppo fosfato da un’altra ATP con formazione del 5-pirofosfomevalonato e liberazione di un’altra molecola di ADP tramite l’enzima fosfomevalonato chinasi. Infinte, tramite l’enzima pirofosfomevalonato decarbossilasi si ha la produzione di un doppio legame nel prodotto (Δ3-isopentenil pirofosfato). L’isomerizzazione di quest’ultimo porta alla formazione del dimetilallil pirofosfato. L’azione della pirofosfomevalonato decarbossilasi agisce in due tappe: dapprima si ha l’aggiunta di un gruppo fosfato proveniente dall’ATP su C3 con formazione dell’intermedio 3-fosfo-5-pirofosfomevalonato e liberazione di ADP; in seguito si ha la decarbossilazione con rimozione di CO2 e fosfato inorganico e formazione dell’isoprene attivato.
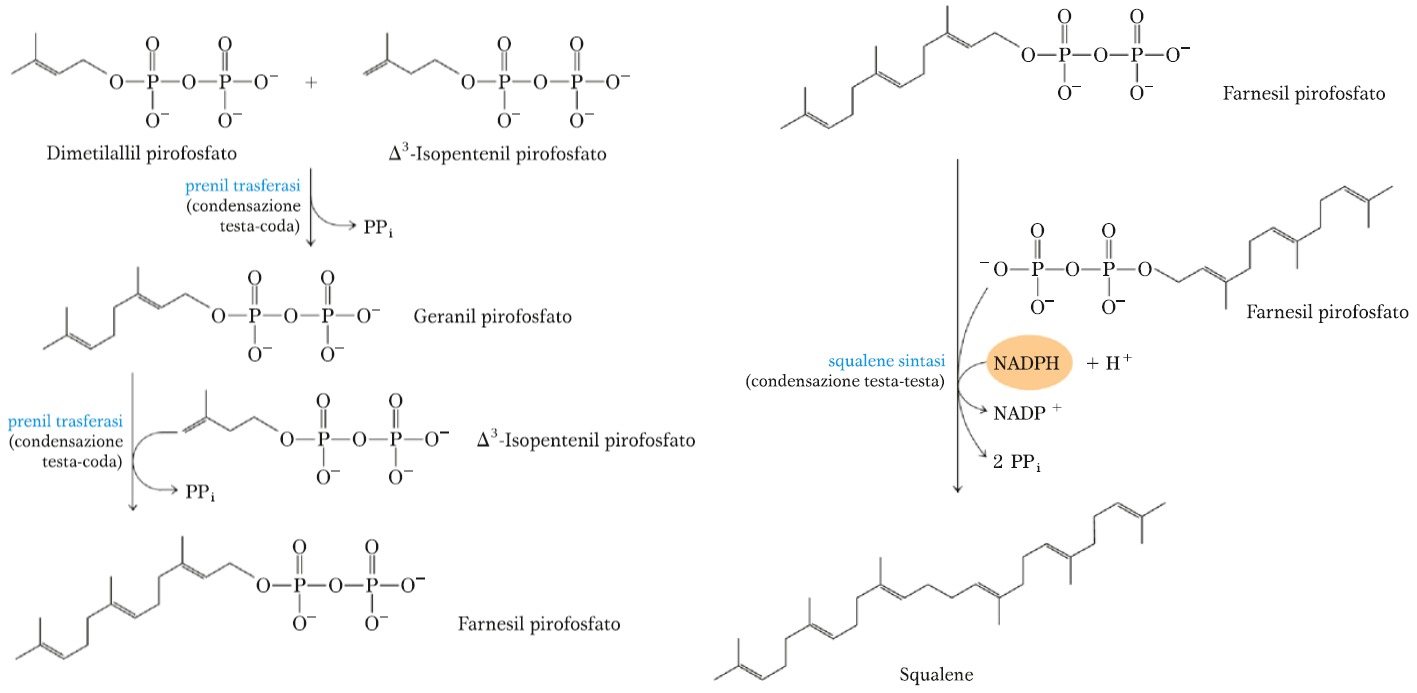
TAPPA 3 – CONDENSAZIONE DI 6 UNITÀ ISOPRENICHE ATTIVATE PER FORMARE LO SQUALENE
L’isopentenil pirofosfato e il dimetilallil pirofosfato vanno incontro ad una condensazione “testa-coda” per formare il geranil pirofosfato (10C) con l’eliminazione di un gruppo pirofosfato (PPi) tramite l’enzima prenil trasferasi. Il geranil pirofosfato subisce un’altra condensazione testa-coda con un Δ3-isopentenil pirofosfato per formare il farnesil pirofosfato (15C) sempre ad opera dell’enzima prenil trasferasi e con eliminazione id un gruppo pirofosfato. Infine, due farnesil pirofosfato condensano testa-testa, eliminando entrambi i gruppi PPi formando lo squalene (30C, 24 nella catena principale e 6 sotto forma di ramificazioni di gruppi metilici) in una reazione catalizzata dall’enzima squalene sintasi che richiede come coenzima NADPH + H+.
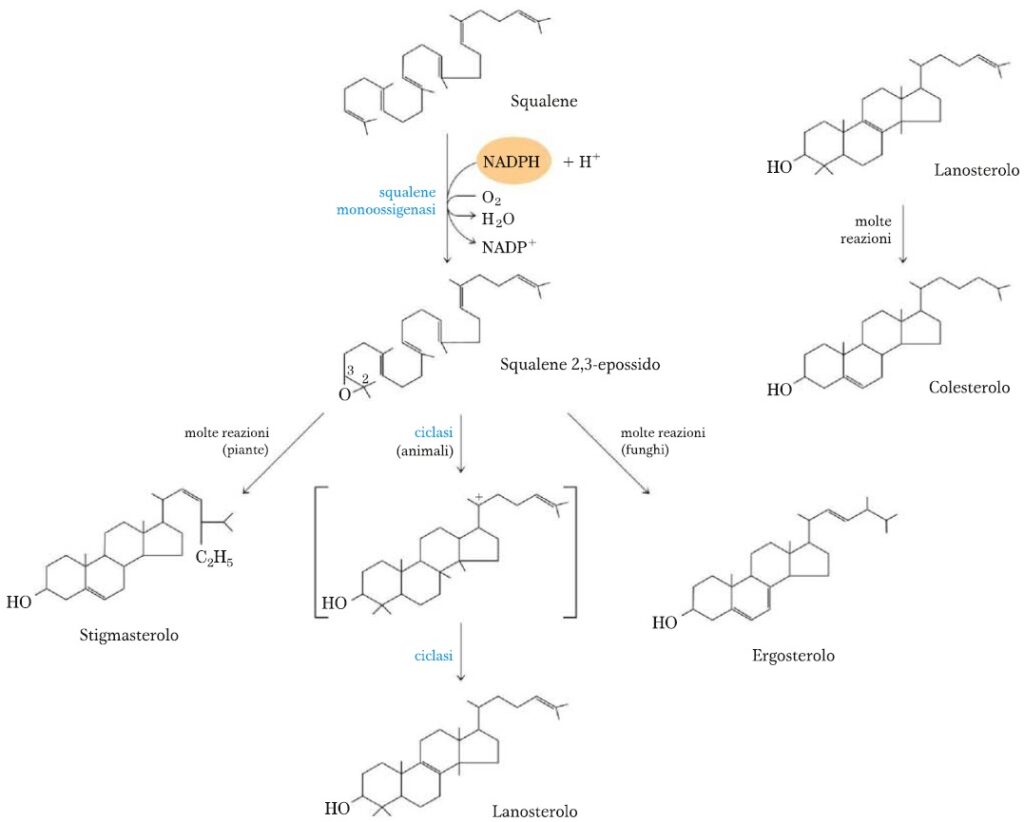
TAPPA 4 – CONVERSIONE DELLO SQUALENE NEL NUCLEO STEROIDEO A 4 ANELLI
Lo squalene monossigenasi aggiunge un atomo di ossigeno (prelevandolo dall’O2) all’estremità dello squalene, formando un epossido (squalene 2,3-epossido). Quest’enzima è un’ossidasi a funzione mista che trasforma in H2O l’altro ossigeno non immesso nello squalene, mediante l’intervento del cofattore NADPH. Nelle cellule animali delle ciclasi formano il lanosterolo che contiene già i 4 anelli caratteristici del colesterolo. Il lanosterolo infine va incontro a circa 20 reazioni (di rimozione e di spostamento di gruppi metilici) che lo trasformeranno in colesterolo.
Il colesterolo può poi andare in contro a diversi destini:
essere incorporato nelle membrane degli epatociti;
essere escreto come acidi biliari: la bile è costituita da acidi biliari e i loro sali, derivati relativamente idrolitici del colesterolo, sintetizzati nel fegato;
esteri del colesterolo: si formano grazie all’enzima acil-CoA-colesterolo aciltrasferasi (ACAT). Questo enzimatrasferisce un acido grasso dal CoA al gruppo ossidrico del colesterolo, convertendolo in una forma ancora più idrofobica. Questi esteri poi sono o conservati nel fegato come gocce lipidiche o trasportati da lipoproteine verso i tessuti che richiedono colesterolo.
La sintesi del colesterolo è regolata in base alla quantità intracellulare di questo, alla concentrazione di ATP e dagli ormoni insulina e glucagone. La tappa di comando è la reazione catalizzata dalla HMG-CoA reduttasi.
La regolazione a breve termine dell’HMG-CoA reduttasi è permessa da una modificazione covalente reversibile, catalizzata dalla proteina chinasi AMP-dipendente (AMPK). Questa, in risposta ad alte concentrazioni di AMP (che implicano scarsa ATP), fosforila (inattivando) l’HMG-CoA reduttasi, impedendo la sintesi del colesterolo.
Anche gli ormoni hanno un entrano in gioco nella regolazione:
glucagone: favorisce la fosforilazione;
insulina: favorisce la defosforilazione.
Questi meccanismi covalenti hanno però un ruolo relativamente meno importante rispetto a quelli che influenzano la sintesi e la degradazione dell’enzima.
La regolazione a lungo termine della via prevede un aumento o una diminuzione della concentrazione di HMG-CoA reduttasi in risposta ai livelli di colesterolo. Il gene che codifica per l’enzima è controllato dalle proteine che legano gli elementi regolatori degli steroli (SREBP). Le SREBP appena sintetizzate vengono immerse nel reticolo endoplasmatico. Quando i livelli di colesterolo e ossisteroli sono alti, le SREBP sono associate nel reticolo con la proteina attizzatrice della scissionedella SREBP (SCAP) che, a sua volta, è ancorata tramite l’interazione con la proteina del gene indotta dall’insulina (INSIG). SCAP e INSIG fungono da recettori per i livelli di steroli. Quando questi sono alti, il complesso SCREBP-SCAP-INSIG è ancorato alla membrana del reticolo endoplasmatico, se invece il livello di steroli decresce, il complesso SCAP-SCREBP viene trasferito al complesso di Golgi, ove due scissioni proteolitiche sulla SREBP rilasciano nel citosol un frammento regolatorio che entra nel nucleo e agisce sui geni che traducono per gli enzimi della sintesi del colesterolo. Quando il livello di steroli aumenta, non si ha più la dissociazione del complesso SREBP-SCAP-INSIG che rimane saldo al reticolo, i proteasomi nel mentre distruggono il frammento regolatorio prima prodotto, portando quindi ad una diminuzione della trascrizione dei geni bersaglio.
La INSIG, inoltre, percepisce alte concentrazioni di colesterolo e favorisce l’attacco dell’ubiquitina all’HMG-CoA reduttasi, portandola alla degradazione da parte dei proteasomi.
Il recettore X del fegato (LXR) è un fattore di trascrizione nucleare attivato da ligandi ossisterolici (che riflettono alti livelli di colesterolo). Se ne conoscono 2 tipi: LXRα (presente solo nel fegato, tessuto adiposo e macrofagi) ed LXRβ (espresso in tutti i tessuti). Quando LXR è legato a steroli, si associa con i recettori X dei retinoidi (RXR). Il dimero così formato va ad attivare la trascrizione di un gruppo di geni che permettono l’abbassamento della concentrazione di colesterolo. Quindi si può dire che SREBP viene attivato da bassi livelli di colesterolo, LXR da alti livelli. Entrambi collaborano per l’omeostasi di questo lipide.
Il colesterolo infine è controllato anche da altri 2 meccanismi regolatori:
alte concentrazioni di colesterolo attivano l’ACAT che incrementa l’esterificazione del lipide per l’immagazinamento;
alte concentrazioni di colesterolo diminuiscono (via SREBP) la trascrizione del gene che codifica per il recettore delle LDL, riducendo la produzione di questo e quindi l’assorbimento del colesterolo dal sangue.
Il colesterolo, che non può essere catabolizzato come combustibile metabolico, quando la somma di quello ottenuto con la dieta supera la quantità necessaria per l’organismo, si ha un accumulo patologico di colesterolo (placche). Queste possono ostruire i vasi sanguigni portando all’aterosclerosi. La formazione delle placche ha inizio quando le LDL aderiscono alla parete dei vasi e vi si accumulano. I macrofagi vengono attratti in queste zone di accumulano ed iniziano a fagocitare LDL fino a danneggiarsi, diventando cellule schiumose e andando in contro ad apoptosi. In un lungo periodo di tempo si formano quindi delle placche costituite da materiale extracellulare, tessuto cicatriziale della muscolatura liscia e cellule schiumose. Queste placche di ateroma possono staccarsi e andare ad occludere un vaso dal lume più piccolo.
Nell’ipercolesterolemia familiare, il recettore per le LDL è difettoso, ne consegue quindi che il colesterolo ematico rimane libero nel sangue, in quanto non può essere più endocitato. Questa situazione è aggravata dalla produzione di colesterolo endogeno, la cui via di sintesi non può essere inibita in quanto il colesterolo ematico non può entrare nel citosol. Una classe di farmaci detti statine influisce proprio sula sintesi del colesterolo, inibendo competitivamente l’HMG-CoA reduttasi, impedendo quindi la sintesi del colesterolo. Questa inibizione è possibile poiché le statine hanno una struttura simile al mevalonato (substrato dell’HMG-CoA reduttasi), con il quale entrano in competizione.








")






")