Le sindromi di Ehlers-Danlos sono un gruppo di patologie clinicamente e geneticamente eterogenee dovuto ad alterazioni nella struttura o sintesi del collagene fibrillare (sono interessati cute, legamenti e articolazioni).
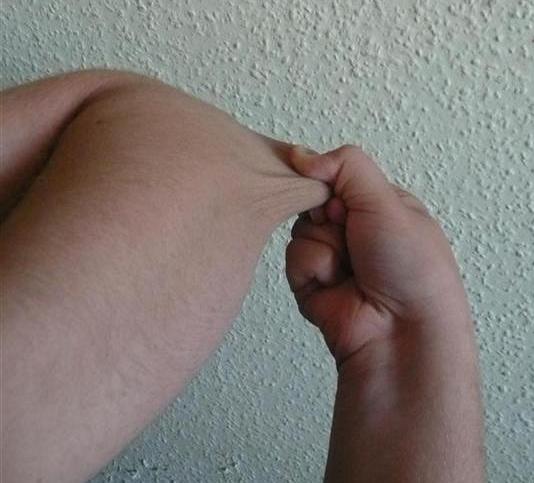
Tra i vari distretti interessati abbiamo la cute: i soggetti presentano cute lassa iperestensibile. Si pensa che i contorsionisti siano affetti da questa sindrome proprio perché in questa sindrome la pelle è iperestensibile e i legamenti e le articolazioni sono ipermobili.
Il collagene è la proteina più abbondante nel regno animale, fornisce la struttura extracellulare di tutti gli organismi multicellulari. Tutti i tipi di collagene sono composti da 3 eliche che contengono la sequenza gly-x-y dove x e y di solito sono prolina e lisina. Attualmente si conoscono 28 tipi di collagene, codificati da 41 geni distribuiti su 14 cromosomi.
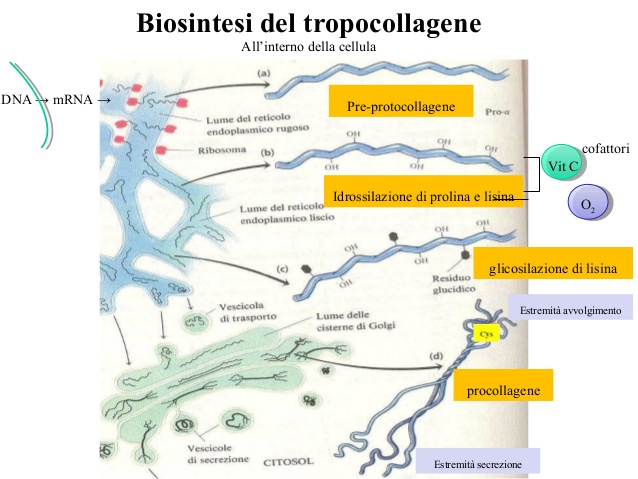
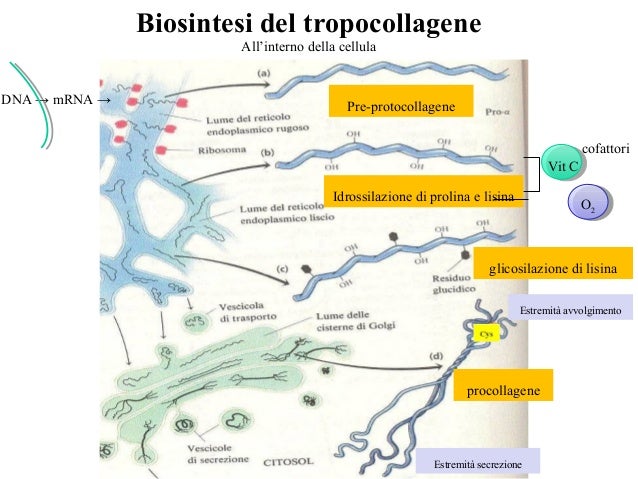
Nel momento in cui il collagene viene sintetizzato, soprattutto da fibroblasti del connettivo o anche da osteoblasti, è una molecola immatura; a seguito di modificazioni biochimiche come idrossilazione di alcuni residui e glicosilazioni, si ha impacchettamento di 3 eliche di collagene, che esce dalla cellula sotto forma di procollagene ancora immaturo. A seguito di tagli a livello delle estremità del collagene da parte di peptidasi si ha il tropocollagene. Fibre di tropocollagene si organizzano a formare legami crociati grazie a enzimi specializzati come la lisin-idrossilasi, legami che coinvolgono l’amminoacido lisina, in tal modo si formano dei fasci paralleli a formare delle vere e proprie fibre di collagene. Inoltre, per la formazione di questi legami crociati sono importanti cofattori come la vitamina C;
Sono state descritte circa 10 sindromi di E.D. di cui le più diffuse sono la tipo 1, 4 e 6:
La sindrome di tipo 1: detta anche tipo gravis, è autosomica dominante, il gene interessato è COL5A (collagene 5), caratterizzata da cute iperestensibile, articolazioni ipermobili e cicatrizzazione cutanea alterata. In questo caso le mutazioni riguardano principalmente la sintesi di collagene che è ridotta oppure mutazioni puntiformi che possono portare alla sintesi di un collagene anomalo;
La sindrome di tipo 4 (tipo arterioso) interessa i vasi. In questo caso le mutazioni possono essere di diverso tipo: ci sono mutazioni che influenzano i livelli di proteina prodotta, esempio mutazioni a livello del promotore, oppure proprio sostituzioni che portano proprio ad un collagene anomalo. È autosomica dominante, il gene è COL3A (collagene 3) ed è caratterizzata da cute sottile e facilmente danneggiata, rotture delle pareti arteriose ed intestinali;
La sindrome di tipo 6 (tipo oculare o cifoscoliotico) comporta una mutazione non riguarda direttamente il collagene ma l’enzima che forma i legami crociati, lisin-idrossilasi, e ricordiamo, che in generale gli enzimi al 50% (cioè quando è mutata una sola copia) riescono a sopperire la mancanza dell’altro 50% e quindi affinché si abbia la patologia è necessario che entrambe le copie del gene siano mutate (cioè in condizione recessiva). È autosomica recessiva, il gene è quindi il gene PLOD della lisina idrossilasi. La caratteristica è la fragilità oculare (distacco retina, rottura cornea) e cutanea.



{kind=link}
{kind=link}








