Il retinoblastoma è un tumore maligno infantile (in genere si sviluppa nei primi 24 mesi) dell’occhio derivato da cellule embrionali della retina, i retinoblasti. Esso può presentarsi in forma ereditaria (40%) con trasmissione autosomica dominante con penetranza incompleta (del 90%) oppure in forma sporadica (60%).
Il retinoblastoma inoltre può essere uni o bilaterale. Quando è unilaterale possono essere presenti molteplici foci separati di cellule neoplastiche (forma multifocale), indicativi della presenza di tumori multipli indipendenti. Le forme bilaterali, così come quelle ereditarie, tendono ad insorgere in età più precoce. Inoltre, i pazienti affetti da forme ereditarie molto spesso hanno tumori bilaterali, mentre solo una piccola frazione dei casi sporadici presenta tumori in entrambi gli occhi oppure multifocali in un singolo occhio.
La presenza di forme ereditarie è indicativa dell’intervento di mutazioni costituzionali che predispongono allo sviluppo del tumore. Lo stesso tipo di mutazioni è coinvolto nell’origine dei tumori sporadici. Tuttavia, il difetto di penetranza indica che la mutazione costituzionale non è sufficiente a far comparire la malattia, ma sono necessari altri eventi per lo sviluppo del retinoblastoma nei soggetti predisposti, seguendo quindi la teoria dei “due colpi” (two hits), o meccanismo “a due tempi”, secondo la quale lo sviluppo del retinoblastoma richiede due eventi mutazionali. Nelle forme ereditarie una prima mutazione è presente in tutte le potenziali cellule bersaglio (i retinoblasti, precursori dell’epitelio differenziato della retina), poiché l’individuo l’ha ereditata per via germinale da uno dei genitori, eterozigote per la stessa mutazione, oppure perché si è verificata una mutazione germinale de novo. Le conseguenze sono uguali nei due casi poiché la mutazione, essendo presente già allo stadio di zigote, farà parte del genotipo costituzionale dell’individuo. Quando in una cellula somatica si verifica il secondo evento mutazionale, con la perdita di eterozigosi e inattivazione dell’allele funzionale, inizia lo sviluppo del tumore per mancata espressione dell’oncosoppressore.
Nelle forme sporadiche entrambe le mutazioni devono avvenire a livello somatico. Perché si sviluppi il retinoblastoma, esse devono essere contemporaneamente presenti nella stessa cellula. Le cellule in cui si è verificata una sola mutazione, così come le loro discendenti, hanno caratteristiche normali; acquisiranno proprietà tumorali solo se e quando comparirà nel loro genoma la seconda mutazione. Questa ipotesi spiega perché il rischio di retinoblastoma è in genere molto più basso negli individui con storia familiare negativa (forme sporadiche) rispetto a coloro che hanno ereditato un gene mutante, per i quali la probabilità di ammalarsi equivale alla penetranza, quindi 0,9 (90%, ovvero 9 individui su 10 portatori del gene manifestano questa malattia). Nei soggetti predisposti basta una sola mutazione in una qualsiasi cellula bersaglio, mentre i tumori sporadici compaiono solo quando si sono verificate indipendentemente due mutazioni, la seconda delle quali deve avvenire in una cellula in cui è già presente la prima. Tale ipotesi spiega anche perché i tumori sono spesso multipli e bilaterali nelle forme ereditarie e perché la loro insorgenza è precoce nelle forme ereditarie.
Caratteristica
Unilaterale
Bilaterale
Frequenza relativa
65%
35%
Età mediana alla diagnosi
22 mesi
11 mesi
Epidemiologia
Generalmente sporadico
Frequente nelle forme ereditarie
Multifocalità
Occasionale
Frequente, in entrambi gli occhi
A livello cromosomico si ha una delezione interstiziale nella banda 14 del braccio lungo delcromosoma 13 (13q14), zona in cui è localizzato il gene del RB. La perdita di un gene (per delezione parziale o completa del cromosoma) comporta l’assenza della proteina prodotta dall’allele deleto. La mancanza di entrambe le copie, che avviene nelle forme con delezioni omozigoti, comporta una completa perdita di funzione del locus.
Questo gene, denominato RB1, è un oncosoppressore, ovvero in condizioni normali ha effetti inibitori sulla trasformazione neoplastica. Il danneggiamento di una singola copia del gene consente ancora il funzionamento del locus e quindi non è associato allo sviluppo del tumore. La perdita della funzione di entrambe le copie determina invece l’inattivazione completa del locus e quindi la comparsa del tumore. La perdita della funzione del locus RB1 può avvenire attraverso diversi meccanismi molecolari. Nelle forme ereditarie il primo evento, costituzionale, è generalmente una mutazione puntiforme, più spesso nonsenso o frame-shift, che comporta la formazione di una proteina prematuramente troncata. Il secondo evento, somatico, è spesso una perdita completa o parziale del cromosoma 13 contenente l’allele RB1 selvatico, a volte accompagnata dalla duplicazione del cromosoma 13 in cui è presente la copia mutata del gene RB1. In altri casi anche la mutazione somatica è puntiforme, all’interno dell’allele RB1 selvatico. Gli stessi tipi di alterazioni, entrambe avvenute a livello somatico, sono presenti nei RB sporadici; quindi, lo stesso gene è coinvolto sia nella sindrome ereditaria che nella genesi della forma sporadica.
In condizioni normali, il gene RB1 è implicato nel controllo del ciclo cellulare. La proteina del retinoblastoma è localizzata prevalentemente nel nucleo dove può essere presente in forma defosforilata (p110RB), che è attiva e lega altre proteine, inibendole, come E2F, che hanno un effetto positivo sulla trascrizione di geni che promuovono la progressione attraverso il ciclo cellulare, o nella forma fosforilata (p112RB), che è inattiva e quindi lascia libere le suddette proteine. La fosforilazione avviene al momento della transizione tra fase G1 e fase S.
Oltre al retinoblastoma, il gene RB1 è inattivato in carcinomi della vescica, della mammella e del polmone.
Un individuo con genotipo affetto da mutazione potrebbe non manifestare il fenotipo malato anche se può trasmettere il gene malato alla generazione successiva (50%). Quindi chiunque erediti un gene RB mutato ha una perdita della funzionalità con aumentato rischio di sviluppare la malattia. Non tutte le persone con la mutazione genica sviluppano la malattia (penetranza incompleta), ma ogni persona con il gene mutato è predisposta a svilupparla, per questo la mutazione ereditaria di RB segue un’ereditarietà autosomica dominante e chiunque eredita il gene esprime un tratto particolare (in questo caso la ridotta funzione di RB e l’aumentato rischio di sviluppo di retinoblastoma).
Clinica
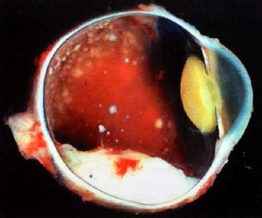
La sintomatologia si presenta generalmente entro i primi 5 anni di vita; il segno principale è un riflesso biancastro del fondo oculare, detto leucocoria, dovuto alla massa tumorale retinica che occupa il corpo vitreo, associata spesso ad una deviazione degli assi oculari, ovvero uno strabismo (dovuto alla perdita di fissazione dell’occhio affetto). Tutto questo porta anche ad una riduzione della visione, dolore e arrossamento degli occhi e ritardo nello sviluppo. Altri segni clinici poco comuni sono l’ipopion (raccolta di essudato infiammatorio purulento nella camera anteriore dell’occhio), l’emorragia del vitreo, il distacco della retina non regmatogeno, il glaucoma neovascolare e la cellulite orbitale (infiammazione dei tessuti oculari dietro il setto orbitale).
Il tumore tende ad espandersi rapidamente verso il vitreo (forma endofitica o intraoculare) oppure verso la coroide e l’uvea (forma esofitica o extraoculare) fino ad interessare i tessuti intorno all’occhio (retinoblastoma orbitale). Il tumore si può anche insinuare nella zona retrooculare fino al nervo ottico, per poi raggiungere il cervello.
Se non viene curato, la dimensione dell’occhio aumenta (buftalmo) e il tumore infierisce su altre parti del corpo, con metastasi al fegato, ai polmoni, linfonodi, midollo osseo e colonna vertebrale (potenzialmente letali).
La forma ereditaria comprende i tumori dovuti a una predisposizione genetica (indipendentemente dall’anamnesi familiare); la maggior parte dei pazienti colpiti da RB ereditario presenta un tumore bilaterale ed è a rischio di sviluppare tumori secondari, in particolare il sarcoma, ma anche il pineoblastoma/tumore soprasellare, il tumore gliale, il melanoma e il carcinoma (vescica, mammella, polmoni).
Diagnosi
In caso di anamnesi familiare positiva, il paziente è sottoposto a visite oculistiche regolari per lo screening del tumore. I bambini che hanno un genitore o un fratello con anamnesi di retinoblastoma devono essere valutati da un oculista poco dopo la nascita e successivamente ogni 4 mesi fino a 4 anni di età.
La diagnosi clinica di retinoblastoma è stabilita con l’esame del fondo oculare utilizzando il RETCAM, che permette di ottenere immagini ad altissima risoluzione e che viene eseguita di norma in anestesia, data l’età del paziente, e con pupille ben dilatate (tramite tropicamide). I tumori appaiono nella retina come rilievi grigio-biancastri singoli o multipli; le origini del tumore possono essere visibili nel vitreo.
Tecniche di imaging possono essere utilizzate per confermare la diagnosi, definire la stadiazione del tumore (dove si trova, in che misura è diffuso, se sta interessando le funzioni di altri organi del corpo ecc.) e determinare se il trattamento è stato efficace. Le indagini possono includere ecografia, tomografia computerizzata (TC) e risonanza magnetica (MRI).
La diagnosi genetica-molecolare è possibile mediante l’identificazione della mutazione del gene RB1. L’analisi citogenetica (cioè dei cromosomi) dei linfociti del sangue periferico è utilizzata per rilevare delezioni o riarrangiamenti che coinvolgono il cromosoma 13 (13q14.1-q14.2). Si raccomanda di proporre le analisi genetiche a tutti i pazienti e ai loro familiari, anche nei casi di retinoblastoma monolaterale.
Terapia
Se il tumore viene trattato quando è ancora intraoculare, più del 90% dei pazienti può essere curato. La prognosi per i pazienti con malattia metastatica è infausta. Nei pazienti con retinoblastoma ereditario l’incidenza del 2o tumore è aumentata; circa il 50% insorge nella zona irradiata. Questi tumori possono comprendere sarcomi e melanomi maligni. Circa il 70% dei pazienti che svilupperà un 2o cancro lo sviluppa entro 30 anni del retinoblastoma primario.
Il trattamento varia a seconda dello stadio di malattia, le dimensioni del tumore, l’età del paziente e la sua clinica. Gli obiettivi sono di eliminare il tumore e salvare la vita del paziente; salvare l’occhio, se possibile; preservare il più possibile la visione ed evitare l’insorgenza di altri tumori, che possono anche essere causati da un trattamento, specie nei bambini con retinoblastoma ereditario.
Nel caso di un piccolo tumore, confinato all’interno dell’occhio, di solito si interviene con:
Terapia laser (fotocoagulazione) nei tumori più piccoli. Durante la terapia, il laser viene utilizzato per distruggere i vasi sanguigni che irrorano la massa neoplastica, attraverso la pupilla. Senza una fonte di ossigeno e nutrienti, le cellule tumorali possono morire.
Termoterapia: con un termolaser che applica calore alle cellule tumorali. Le temperature non sono elevate come quelle utilizzate nella fotocoagulazione ed alcuni dei vasi sanguigni sulla retina possono essere risparmiati.
Congelamento del tumore (crioterapia) utilizzando l’azoto liquido, per congelare ed uccidere le cellule anomale. Il medico utilizza una piccola sonda di metallo, la quale viene collocata all’interno o vicino alla massa neoplastica, prima di essere raffreddata. Questo processo di congelamento e scongelamento, ripetuto un paio di volte in ogni sessione di crioterapia, induce la morte delle cellule cancerose. La procedura è più efficace quando il tumore è di piccole dimensioni e localizzato solo in alcune parti dell’occhio (soprattutto nella parte anteriore).
Le masse tumorali più estese possono essere trattate con uno dei seguenti trattamenti, spesso in combinazione tra loro:
Chemioterapia utilizzando agenti chimici, come la ciclofosfamide per ridurre i tumori nell’occhio o per eliminare eventuali cellule tumorali residue dopo altri trattamenti. I farmaci chemioterapici possono essere somministrati per via orale, endovenosa o intra-arteriosa. La chemioterapia sistemica può essere usata anche per trattare un retinoblastoma diffuso ai tessuti oltre il bulbo oculare o ad altre zone del corpo. La chemioterapia intra-arteriosa prevede l’infusione del chemioterapico direttamente nell’arteria oftalmica, che viene raggiunta attraverso un micro-catetere inserito a livello inguinale nell’arteria femorale; insieme alla chemioterapia intra-vitreale, garantisce indiscutibili vantaggi in termini di maggiore efficacia e minore tossicità sistemica.
Radioterapia che utilizza raggi ad alta energia per uccidere le cellule tumorali o rallentarne il tasso di crescita.
Radioterapiaesterna, concentrando fasci di radiazioni ad alta potenza generate da una sorgente all’esterno del corpo. Attualmente si preferisce evitare la radioterapia esterna per evitare il rischio di effetti collaterali, come l’insorgenza di tumori secondari nelle regioni irradiate.
Radioterapia interna (brachiterapia). La procedura è possibile se il tumore non è troppo grande. Durante la radiazione interna, il dispositivo di trattamento, che consiste in un piccolo disco di materiale radioattivo, viene temporaneamente collocato in prossimità del retinoblastoma. La placca radioattiva è suturata sopra le cellule cancerose e lasciata in sede per alcuni giorni, durante i quali somministra lentamente le radiazioni al tumore. L’applicazione vicino al retinoblastoma riduce la possibilità che il trattamento possa produrre effetti sul tessuto sano dell’occhio.
Chirurgia: quando il tumore è troppo esteso per essere trattato con altri metodi. I pazienti con un retinoblastoma unilaterale possono essere sottoposti alla chirurgia di enucleazione per rimuovere l’occhio malato. Sono quindi utilizzate delle protesi (occhio artificiale) per riempire la zona occupata dal bulbo oculare. I bambini che perdono la vista in uno dei loro occhi sono, di solito, in grado di adattarsi rapidamente al cambiamento. I pazienti con retinoblastoma bilaterale possono subire l’enucleazione se la visione non può essere preservata con altri trattamenti.









")


")
")

")