Le distrofie muscolari sono malattie che coinvolgono diversi geni, ma il più frequente è il gene della distrofina, uno dei geni più grossi dell’organismo, contiene circa 79 esoni, ed è anche un gene molto suscettibile a mutazioni.
La patogenesi è caratterizzata dal fatto che a seguito dell’indebolimento del complesso distrofina-glicoproteine-citoscheletro, tra le altre cose si ha rottura della membrana con entrata di calcio e perdita dell’osmosi cellulare che porta alla morte della cellula.
Sono un gruppo eterogeneo di malattie ereditarie caratterizzate da debolezza muscolare e atrofia a carattere ingravescente. Riguardano mutazioni in geni che codificano componenti del complesso distrofina-glicoproteine: indebolimento del sarcolemma, rottura della membrana, morte della fibra muscolare, degenerazione muscolare progressiva, debolezza muscolare, insufficienza respiratoria e cardiaca. Ne esistono vari tipi, le principali sono le distrofie muscolari di Duchenne e di Becker.
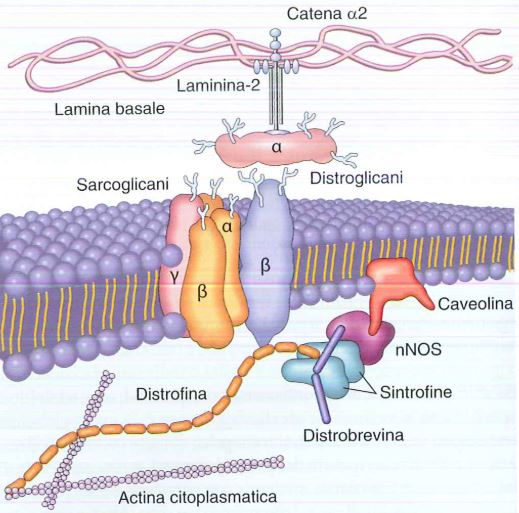
Il complesso distrofina-glicoproteine è importante per il collegamento citoscheletro-matrice e per la stabilizzazione della membrana durante i cicli contrazione-rilassamento. Il collegamento impedisce fratture del sarcolemma indotte da sforzo meccanico durante la contrazione delle cellule muscolari. Nel momento in cui la distrofina manca o non funziona correttamente è molto più probabile che in seguito allo sforzo muscolare vi sia una lacerazione delle fibre e quindi danno a livello della membrana dei miociti con ingresso di calcio che induce la necrosi di queste cellule che vanno incontro a morte progressiva. Il calcio è circa 10.000 volte più concentrato all’esterno della cellula e questo è importante perché l’entrata di calcio in cellula stimola tutta una serie di processi come l’attivazione di enzimi quali le fosfolipasi che degradano membrana cellulare, le proteasi che degradano altre proteine ed enzimi che degradano DNA che poi portano alla distruzione della cellula stessa. Quindi la cellula va incontro a morte per necrosi.
La distrofina, una proteina intracellulare forma una interfaccia tra le proteine del citoscheletro e un gruppo di proteine transmembrana, i distroglicani e i sarcoglicani. Le proteine citoscheletriche sono l’actina citoplasmatica, la distrobrevina e le sintrofine (queste due formano un legame con l’ossido di azoto sintetasi di tipo neuronale (nNOS) e con la caveolina (ancorata sul versante citosolico della membrana). Le proteine transmembrana interagiscono con elementi matrice extra tra cui la laminina. Mutazioni di sarcoglicani, caveolina porta a distrofie autosomiche dei cingoli. Mutazioni di laminina comporta distrofie congenite. Mutazioni alla distrofina portano a distrofia muscolare di Duchenne.
Il gene colpito per le distrofinopatie è quindi la distrofina, 79 esoni su 2.5 Mb (trascritto: 14 Kb), gene presente sul cromosoma X e quindi colpisce più i maschi rispetto alle femmine (incidenza 1:3.500 maschi) e in maniera grave. Questo perché nel momento in cui i maschi ereditano il gene mutato manifestano necessariamente la patologia, mentre nelle femmine si ha manifestazione della patologia a seconda di quale cromosoma X viene inattivato per effetto della lyonizzazione; chiaramente se viene inattivato l’X dove è presente il gene mutato, la malattia non c’è, se invece viene inattivato il cromosoma sano la malattia si manifesta.
La malattia è caratterizzata da eterogeneità allelica e quindi ha gravità diversa a seconda della mutazione che caratterizza il gene della distrofina. Nel 30% dei casi vi è un’anamnesi familiare negativa (mutazione de novo).
Vi si distinguono prevalentemente due tipi di distrofinopatie:
distrofia muscolare di Duchenne (DMD): delezioni che impediscono sintesi proteica, quadro clinico grave e insorgenza precoce;
distrofia muscolare di Becker (BMD): sintesi di proteine anomale con funzionalità ridotta (con amminoacidi diversi o che mancano di alcune porzioni), quadro clinico meno grave di DMD, insorgenza più tardiva;
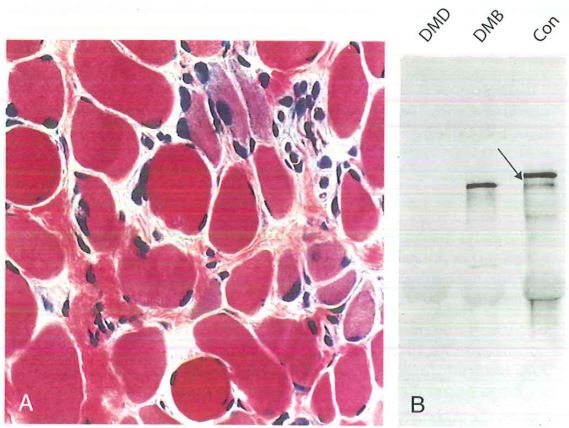
A:Distrofia muscolare di Duchenne (DMD) che mostra variazione nelle dimensioni delle fibre muscolari, aumento del tessuto connettivo endomisiale e fibre in rigenerazione (colore blu). B:Western blot che mostra assenza di distrofina nella DMD e alterazione nelle dimensioni della distrofina nella distrofia muscolare di Becker (DMB), a confronto con un soggetto normale di controllo (freccia) (Con). (Per gentile concessione di Dr. L. Kunkel, Children’s Hospital, Boston, MA). Fonte: Le basi patologiche delle malattie.
Per quanto riguarda la Duchenne l’insorgenza varia tra i 3 e i 5 anni. I bambini colpiti hanno difficoltà a deambulare o ad alzarsi da terra e mostrano una degenerazione dei muscoli della coscia e del bacino. La malattia diventa molto grave già entro i 10/15 anni di età quando i soggetti non sono già più in grado di camminare. Un enzima importante che si dosa già quando si fanno le normali analisi del sangue è la creatinina fosfochinasi (CPK), un enzima che serve per l’energia del muscolo, il cui aumento nel sangue è indice di danno muscolare proprio perché quando le cellule muscolari hanno un danno di membrana liberano questa proteina in circolo. In genere questi pazienti muoiono intorno ai 20 anni per indebolimento della muscolatura soprattutto cardiaca ma anche polmonare andando incontro a insufficienza respiratoria.
Per quanto riguarda invece la Distrofia di Becker, è più rara con un’incidenza di 1/20.000 circa maschi. Questo tipo di patologia provoca debolezza e atrofia nella muscolatura ma l’esordio è tardivo e la capacità di camminare viene persa più tardi intorno ai 25/30 anni. Dunque, comunque la patologia si manifesta, ma più tardivamente, e i pazienti possono vivere sino ai 50 anni. La morte sopraggiunge sempre per insufficienza respiratoria, l’interessamento cardiaco è meno frequente e le facoltà intellettive sono quasi sempre normali. La distrofia di Becker è più rara della Duchenne e il motivo sta sempre nel tipo di mutazione.
Mutazioni delle distrofie muscolari di Becker e Duchenne sono:
delezione: il 65% dei maschi affetti da DMD hanno delezioni del gene per la distrofina che possono interessare uno o più esoni:
delezione frameshift con spostamento della cornice di lettura che porta ad un troncamento della proteina che è instabile e viene degradata (Duchenne) per cui il fenotipo è grave;
delezioni in frame (delezioni di 3 e multipli) con mantenimento della cornice che porta ad una proteina parzialmente funzionante, priva di un segmento interno (Becker);
mutazioni di siti di splicing: anche qui tutto dipende se la cornice di lettura viene mantenuta o meno. Avvengono nel 30% dei casi;
duplicazione di esoni: anche qui dipende se la cornice viene mantenuta. 5% dei casi. La probabilità che la cornice di lettura venga mantenuta è più bassa rispetto a quella in cui la cornice shifta e infatti la Duchenne è più frequente della Becker.
mutazioni puntiformi come la sostituzione di C3304Y (cisteina scambiata con una tirosina): tale mutazione impedisce alla distrofina di legarsi al β-destroglicano, altra proteina del citoscheletro. Tale mutazione ha effetti minori sulla malattia.
Per trattare i pazienti con DMD si è cercato di sfruttare la terapia genica utilizzando vettori virali come retrovirus e lentivirus derivati da HIV (i lentivirus fanno parte della famiglia dei retrovirus) e adenovirus; questi vettori sono stati modificati in laboratorio e usati per infettare cellule muscolari per veicolare il gene sano e correggere il DNA. Questo è stato un successo nei topi transgenici ma la sperimentazione clinica nell’uomo non è ancora iniziata perché si devono trovare mezzi efficaci di accesso alle cellule muscolari, inoltre la lunghezza del gene per la distrofina (14 Kb) è maggiore della capacità di incorporazione di un adenovirus. Infine sono stati usati anche retrovirus, ma poiché questi infettano solo cellule in attiva proliferazione, il gene è stato introdotto in mioblasti embrionali e i mioblasti trapiantati in vitro esprimono la distrofina solo transitoriamente.







")





")
