La macroglobulinemia di Waldenström è una rara neoplasia monoclonale che coinvolge principalmente le cellule B e corrisponde all’entità clinica del linfoma linfoplasmocitico.
Colpisce principalmente gli individui di età superiore ai 60 anni, con una leggera predominanza nei maschi.
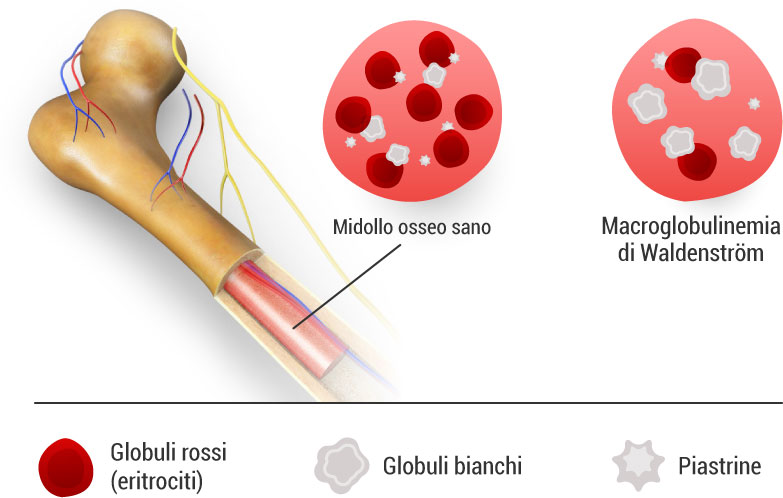
La macroglobulinemia di Waldenström è caratterizzata dalla crescita incontrollata di cellule B e plasmacellule. Queste cellule infiltrano il midollo osseo, contribuendo a costituire oltre il 10% della cellularità midollare.
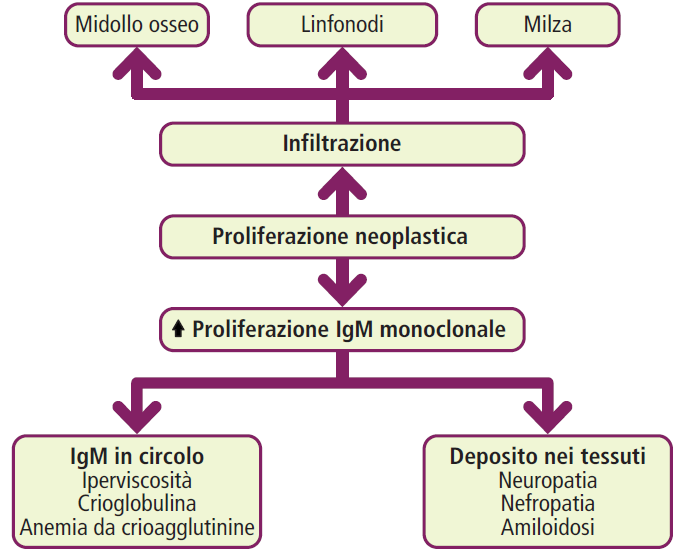
Nel plasma, si osserva la presenza di una “componente M” (monoclonale) dovuta all’iperproduzione di una gammaglobulina. In questo caso, la classe di immunoglobuline coinvolta è l’IgM.
La malattia è associata a una mutazione del gene MYD88, che è presente in circa l’85-95% dei casi. Questa mutazione è coinvolta nella segnalazione del recettore Toll-like e contribuisce alla proliferazione delle cellule B.
A differenza del mieloma multiplo, nella macroglobulinemia di Waldenström mancano le lesioni osteolitiche (danni alle ossa). La manifestazione clinica predominante è la sindrome da iperviscosità, causata dall’elevata concentrazione di IgM nel sangue (> 3 g/dl).
Per lungo tempo, la macroglobulinemia di Waldenström è stata considerata una variante del mieloma multiplo a causa delle somiglianze tra le due condizioni. Tuttavia, attualmente l’Organizzazione Mondiale della Sanità la classifica tra i linfomi a basso grado di malignità. Questo significa che, sebbene sia una neoplasia, ha un comportamento meno aggressivo rispetto ad alcuni altri tipi di linfomi.

Manifestazioni Cliniche
Da un punto di vista clinico la macroglobulinemia è asintomatica in molti casi e per molti anni durante il suo progredire.
Le manifestazioni cliniche includono:
- sindrome da iperviscosità: vertigini, cefalea, disturbi visivi e astenia dovuti alla viscosità del sangue;
- epatomegalia (ingrossamento del fegato), linfoadenopatia (ingrossamento dei linfonodi) e occasionali emorragie;
- amiloidosi: un accumulo di porzioni di catene leggere di immunoglobulina monoclonale in vari organi sotto forma di una proteina anomala detta amiloide, che porta alla progressiva insufficienza d’organo e può colpire cuore, nervi periferici, reni, tessuti molli, fegato e polmoni;
- anemia o trombocitopenia: rispettivamente, basso numero di globuli rossi e piastrine causato da una diffusa infiltrazione midollare e soppressione della normale produzione di cellule del sangue;
- fenomeni autoimmuni: malattia da agglutinine fredde, possibile neuropatia periferica.
Spesso il soggetto colpito si dimostra più suscettibile alle infezioni, fino alla setticemia.
In alcuni casi è possibile che le cellule tumorali infiltrino i polmoni producendo masse o versamenti pleurici (fluido che si accumula nel torace), mentre sono invece rari le lesioni ossee e l’interessamento renale.

Diagnosi
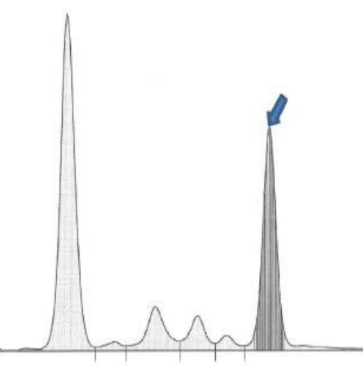
La diagnosi si basa su esami ematochimici, elettroforesi sierica (per identificare il picco monoclonale di IgM) e biopsia del midollo osseo che conferma l’infiltrazione di cellule B e plasmacellule.
Altri esami utili possono essere la tomografia computerizzata di torace/addome/pelvi per determinare l’entità dell’organomegalia, nei pazienti con patologia sintomatica candidati a trattamento e l’analisi citogenetica o l’ibridazione in situ a fluorescenza (FISH), per valutare eventuali anomalie cromosomiche.
Trattamento
Il trattamento è spesso basato sulla gravità dei sintomi e sulla presenza di complicanze.
In caso di malattia sintomatica è prevista come prima linea di terapia una chemio-immunoterapia (ciclofosfamide + desametasone + rituximab oppure bendamustina + rituximab).

La plasmaferesi può essere utilizzata come supporto per ridurre l’iperproduzione di IgM e migliorare la viscosità del sangue.
Di recente, come trattamento di seconda linea è stato approvato Ibrutinib, che agisce come inibitore delle proteine chinasi.
Nei casi più gravi, e comunque quando il paziente è compatibile, si procede con un trapianto di midollo osseo.
La sopravvivenza varia, ma con i trattamenti attuali, molti pazienti possono vivere a lungo con una buona qualità di vita. In assenza di sintomi, i pazienti affetti da macroglobulinemia di Waldenström possono condurre una vita pressoché normale, sottoponendosi a regolari controlli per il monitoraggio della malattia.
Fonte: JassenConTe; Wikipedia.