
Fonte: Le basi patologiche delle malattie.
È la più comune patologia genetica grave nelle popolazioni caucasiche. Autosomica recessiva con incidenza di circa 1/2.500 nati e 1/20.000 in asiatici e africani. Anche le mutazioni che determinano la patologia sono tipiche di alcune aree e addirittura tipiche di alcune regioni, per esempio in Sardegna ci sono mutazioni tipiche della regione. È caratterizzata da affezioni da eccesso di secrezioni a livello di tutte le ghiandole esocrine con livello di gravità variabile nei diversi organismi.
Effetti più gravi coinvolgono polmoni (ostruzioni, infezioni, infiammazioni) e pancreas (insufficienza del pancreas esocrino.
Prognosi determinata dalle affezioni polmonari: mortalità per insufficienza respiratoria nel 90% dei casi. Attesa di vita 40 anni (era 10 anni nel 1960).
Tali pazienti sono molto suscettibili a infezione da patogeni che infettano i polmoni come pseudomonas aeruginosa (patogeno normalmente opportunista). Infatti, a causa dell’alto rischio di infezioni, questi pazienti hanno bisogno di vivere in ambienti quasi sterili e far uso di antibiotici e altri medicinali per aumentare l’attesa di vita.

CFTR
Da un punto di vista genetico, il gene coinvolto è CFTR (Cystic Fibrosis Transmembrane Regulator, 7q31.2) identificato nel 1984 e clonato nel 1989. La proteina codificata dal gene è formata da 1480 amminoacidi ed è un canale del cloro regolato da cAMP, che regola il trasporto di Cl– e indirettamente di Na+ negli epiteli e di conseguenza di acqua.
In basso, CFTR dal gene alla proteina. La mutazione più comune nel gene CFTR induce un deficit di ripiegamento proteico nel reticolo endoplasmatico/Golgi e la degradazione di CFTR prima che raggiunga la superficie cellulare. Altre mutazioni agiscono sulla sintesi di CFTR, NBD, sul dominio R e sui domini di membrana.
Fonte: Le basi patologiche delle malattie.
Il gene CFTR codifica per un canale transmembrana costituito da due domini transmembrana ognuno costituito da 6 alfa eliche. Ognuno dei due domini è connesso ad un dominio citoplasmatico legante nucleotidi come ATP, chiamato Nucleotide Binding Domain (NBD). I nucleotidi sono importanti perché tale canale è regolato da cAMP. Inoltre è presente un dominio regolatore (R) che regola l’apertura e chiusura del canale grazie al fatto che contiene siti di fosforilazione delle protein-chinasi A e C. L’attivazione del canale CFTR è mediata da un agonista che induce un aumento di cAMP seguito dall’attivazione di una protein-chinasi A che fosforila il dominio R. A seguito di questo segnale, il dominio NDB idrolizza l’ATP che tiene legato e questo è essenziale per l’apertura e chiusura del canale.
Si hanno mutazioni nel 3% della popolazione caucasica. Nel 70% vi è la delezione di un solo amminoacido, la fenilalanina in posizione 508 (il frame viene mantenuto): tale mutazione fa sì che il canale non arrivi in membrana e quindi le cellule ne sono prive (1.400 mutazioni diverse nel restante 30% dei casi).
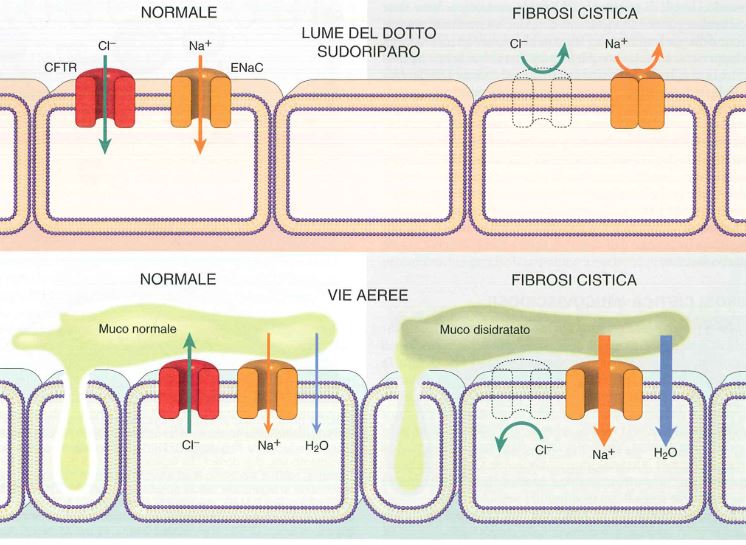
Normalmente il CFTR è un canale che permette il passaggio del cloro all’esterno della cellula delle ghiandole esocrine dell’epitelio di rivestimento del tratto respiratorio, gastrointestinale e riproduttivo e regola negativamente il canale ENaC (canale epiteliale del sodio) e quindi normalmente entra poco sodio in cellula e viene mantenuta l’omeostasi.
In fibrosi cistica non vi è CFTR e quindi vi è ridotta escrezione di Cl–, inoltre non è più regolato ENaC e quindi vi è un iperassorbimento di sodio e per osmosi anche di acqua. Questo porta ad un assottigliamento del liquido di superficie presente sulle cellule ciliate dei polmoni e fa sì che tali ciglia non funzionino più correttamente e non possano rimuovere i microrganismi.
Nelle ghiandole sudoripare, invece, il CFTR normalmente permette il riassorbimento di cloro in cellula e in questo caso se CFTR è mutato, ENaC funziona di meno: se CFTR non c’è, il cloro non può entrare in cellula, ENaC funziona di meno e quindi non può più entrare Na+ e perciò avremo alta concentrazione di sodio e cloro all’esterno cellula, fenomeno che spiega il sudore molto salato.
Fonte: Le basi patologiche delle malattie.
Classe di mutazioni

- Classe 1: produzione difettiva. Sono mutazioni che inseriscono codoni di stop o frame shift per cui il canale non viene prodotto ed il fenotipo è grave;
- Classe 2: processamento difettivo. A queste appartengono le mutazioni ΔF508 che rappresentano il 70% dei casi: tale mutazione fa sì che la proteina venga riconosciuta come anomala a livello del Golgi e distrutta attraverso il sistema ubiquitina-proteasoma. Anche in questo caso abbiamo assenza di proteina su membrana e il fenotipo è grave;
- Classe 3: regolazione difettiva. Le mutazioni riguardano il dominio regolatore del CFTR. In questo caso il canale arriva in membrana ma non funziona correttamente, perciò abbiamo fenotipi intermedi perché dipende da quanto sia difettato il canale;
- Classe 4: conduttanza difettiva. La mutazione riguarda la struttura interna delle alfa eliche;
- Classe 5: produzione ridotta. Il canale arriva in membrana ma la produzione è ridotta. In questo caso sono mutazioni che interessano il promotore, il fenotipo è meno grave perché comunque c’è una quota di proteina C;
- Classe 6: degradazione accelerata. Il CFTR arriva in membrana ma il suo turnover di vita/morte è accelerato e quindi il canale rimane poco tempo in membrana perché viene subito degradato.

Caratteristiche cliniche della malattia
Fonte: Le basi patologiche delle malattie.
Nell’apparato respiratorio il muco spesso e viscoso ostruisce le vie respiratorie e rallenta la rimozione di virus e batteri provocando difficoltà respiratorie e infezioni polmonari ricorrenti. Inoltre si possono sviluppare dei polipi nasali.
A livello del pancreas il muco ostruisce i dotti che portano gli enzimi digestivi dal pancreas all’intestino. Ne consegue malassorbimento degli alimenti e crescita corporea rallentata.
Nell’apparato riproduttivo si ha ostruzione dei vasi deferenti e infertilità nel 95% dei casi. Anche nelle femmine la fertilità è ridotta.
Per quanto riguarda la pelle si ha malfunzionamento delle ghiandole sudoripare che rilasciano elevate quantità di NaCl nel sudore (sudore salato).
Alla nascita può esserci l’ileo del meconio (ostruzione intestinale): con il temine meconio ci si riferisce alle prime feci che vengono eliminate dai neonati di solito entro 12/24 ore dalla nascita. Nella fibrosi cistica a causa della formazione di tappi di muco denso e viscoso in tenue, il meconio non può essere eliminato e questa condizione si chiama proprio ileo da meconio.

Diagnosi
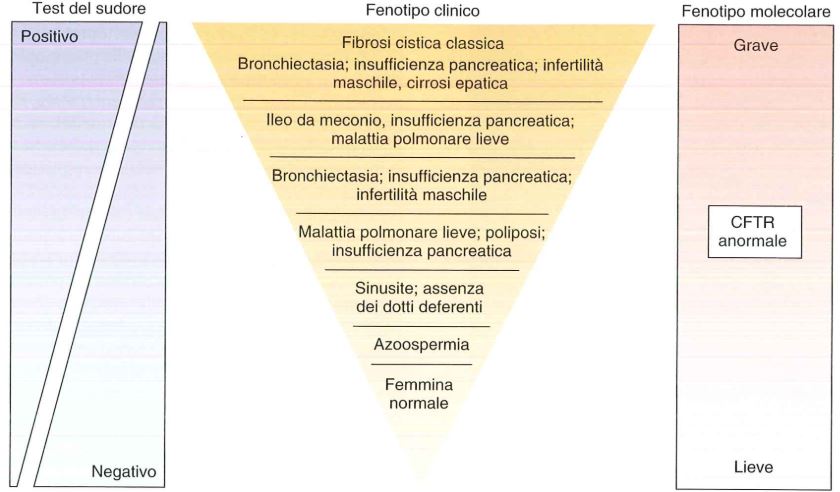
Prima del 1989 si valutavano criteri clinici e test del sudore: elevate concentrazioni di NaCl in sudore erano indicative della patologia. Dopo 1989, oltre a misure funzionali (test del sudore e potenziali nasali), grazie alla scoperta della sequenza sono state possibili anche genotipizzazioni di mutazioni e screening di mutazioni neonatali (cioè viene fatto lo screening alla nascita).
Inoltre, la misurazione della differenza dei potenziali trans-nasali in vivo può essere un utile test aggiuntivo. Individui con fibrosi cistica presentano una differenza di potenziale transepiteliale in vivo significativamente inferiore rispetto ai controlli.
Con lo screening neonatale si va a valutare su campione di sangue ottenuto dal tallone al 2°-4° giorno della nascita la presenza del tripsinogeno immunoreattivo – ImmunoReactive Trypsinogen (IRT), proteina molto presente nei pazienti affetti. Se IRT è positivo, si procede con uno screening genotipico andando a valutare le mutazioni: se positivo per due mutazioni è possibile diagnosticare fibrosi cistica (ricordiamo che la malattia è recessiva); se positivo per una sola mutazione (quindi su un solo allele) si fa un test del sudore per capire se abbiamo effettivamente un paziente in eterozigosi portatore oppure se il paziente ha la fibrosi cistica.
Facendo il test del sudore, se la concentrazione di NaCl è >60 mmol/L vi è diagnosi di fibrosi cistica, se la concentrazione è <40 mmol/L, il paziente è portatore, se compreso tra 40 e 60 l’individuo è border-line e bisogna fare analisi più approfondite.
NB.: basta una quantità del 10% di CFTR funzionante perché non si abbia la malattia. Sotto il 10% cominciano a comparire i primi sintomi. L’insufficienza pancreatica compare quando i livelli di CFTR sono <1%.
L’analisi genetica di 1° livello consiste nella ricerca delle mutazioni più frequenti (delezione di fenilalanina) nella popolazione mediante un immunoblotting con sonde marcate.
L’analisi di 2° livello utilizza sistemi di scanning (sequenziamento) di tutti gli esoni e delle regioni di splicing del gene CFTR (è più approfondito del 1° livello): conferma il 1° livello e in più trova altre mutazioni che non rientrano nel 1° livello.
L’analisi di 3°livello cerca di individuare le mutazioni nel 10-15 % dei soggetti in cui le precedenti analisi non hanno avuto successo: ricerca di inserzioni e/o delezioni.
Trattamento CF

Per il trattamento di insufficienza pancreatica si somministrano enzimi pancreatici ai pasti.
Per i problemi di assorbimento degli alimenti si aumenta l’apporto di calorie.
Per le manifestazioni polmonari si effettuano vaccini contro alcuni patogeni come pseudomonas aeruginosa, si applica profilassi e terapia antibiotica, fisioterapia, farmaci broncodilatatori, corticosteroidi orali, nei casi più gravi trapianto di polmone.
Inoltre si sta tentando di riuscire nella terapia genica cercando un modo per veicolare il gene CFTR nei distretti interessati. Il problema è riuscire a veicolare un gene molto grande, farlo esprimere nella corretta maniera, nella giusta quantità e non avere un’espressione transitoria ma che sia prolungata nel tempo. Al momento sono terapie in studio, ultimamente si sta cercando di far esprimere il gene corretto alle cellule staminali e poi impiantarle per cercare di curare la patologia.