Sono malattie mendeliane autosomiche recessive caratterizzate da una mancanza o diminuzione della sintesi della catena α o β dell’emoglobina. La β-talassemia (beta-talassemia) è la più diffusa.
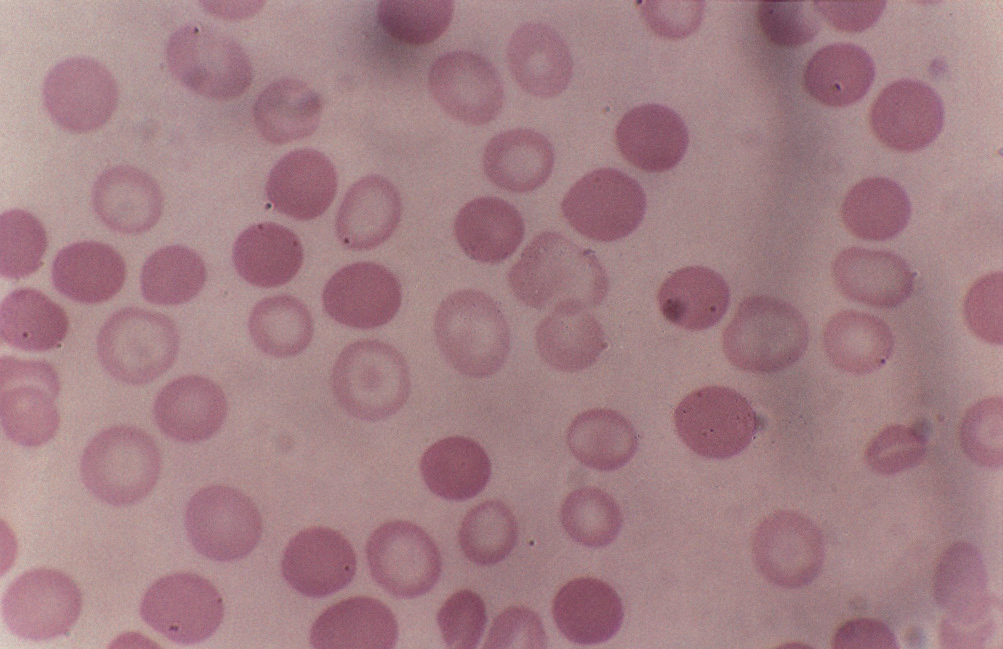
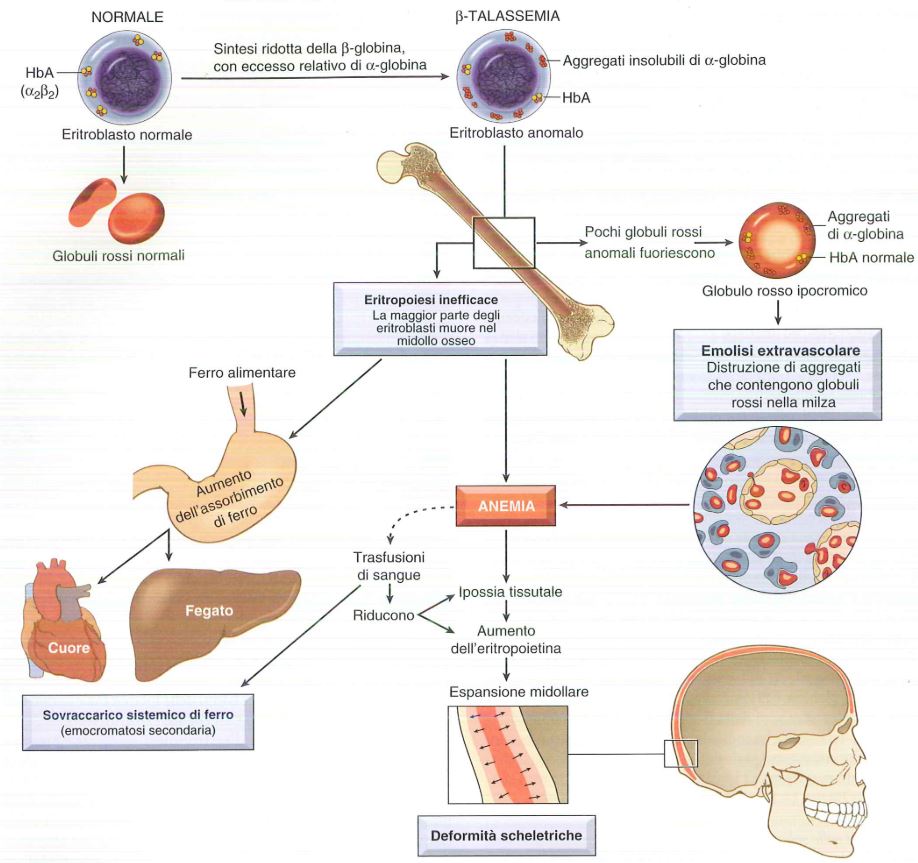
Le conseguenze ematoliche derivano dal basso livello di emoglobina (Hb) intracellulare (ipocromia) ma anche dal relativo eccesso di catene difettive (nella β-talassemia si accumulano le catene α in eccesso formando inclusioni insolubili con danno alla membrana). Molti precursori dei globuli rossi muoiono per il danno alla membrana e vanno incontro ad apoptosi.

Nella β-talassemia grave la maggior parte dei precursori dei globuli rossi fa questa fine e questo porta ad eritropoiesi inefficacie. Inoltre i globuli rossi che vengono rilasciati dal midollo osseo presentano inclusioni e danni alla membrana e hanno una tendenza al sequestro splenico e all’emolisi extravascolare in 20 giorni.
Le catene β sono codificate da un gene localizzato sul cromosoma 11.
Ci sono diversi tipi di β-talassemie:
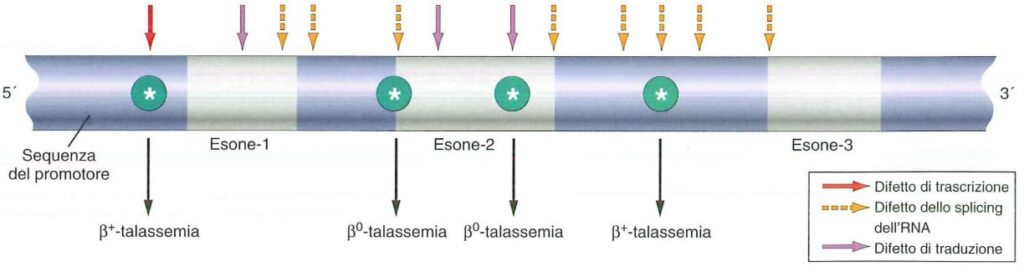
- beta zero β0: causate da mutazioni frame shift o mutazioni che inseriscono codoni stop oppure splicing anomali che spostano la cornice di lettura. In questo caso vi è totale assenza di catene β;
- beta più β+: in questo caso si ha ridotta sintesi e quindi le mutazioni riguardano il promotore e i fattori di trascrizione si legano poco o non si legano affatto;
Il gene della β-globina è un punto caldo di mutazioni nel senso che sono state identificate moltissime mutazioni a carico di questo gene, più di 100 mutazioni puntiformi, in genere sostituzioni. Queste mutazioni possono interessare il promotore causando β+, mutazioni stop codon o frame shift che determinano una interruzione della catena causano β0, e mutazioni di splicing che riguardano per lo più esoni ed introni. Raramente ci sono delezioni (più frequenti nell’α-talassemia).
Le mutazioni di splicing possono portare a β0 nel momento in cui il frame viene perso, oppure β+ se il frame viene mantenuto, e in questo caso avremo emoglobine anomale.
Fonte: Le basi patologiche delle malattie.

Patogenesi beta-talassemie
Fonte: Le basi patologiche delle malattie.
Nelle β-talassemie i globuli rossi sono più piccoli e ipocromici che tendono ad andare in apoptosi perché c’è la precipitazione delle catene α in eccesso che causa danno alla membrana. Questa è un’anemia con condizioni di ipossia perché naturalmente la quota di ossigeno trasportata è bassa, quindi i pazienti sono costretti a fare continue trasfusioni che portano ad un sovraccarico di ferro in organi parenchimali come il fegato.
L’assorbimento del ferro alterato favorisce l’accumulo di ferro in organi parenchimali. L’assorbimento del ferro avviene nel duodeno ed il livello dipende dal contenuto corporeo totale di ferro e dall’eritropoiesi, o più specificatamente dalla richiesta nei precursori eritroidi. Non appena i depositi aumentano, l’assorbimento del ferro si riduce e viceversa. Nel caso della β-talassemia l’assorbimento di ferro è aumentato nonostante l’eccesso di riserve nell’organismo perché si abbassano i livelli di epcidina, un regolatore negativo dell’assorbimento del ferro. Questi soggetti, poi, facendo delle trasfusioni sono sovracaricati di ferro, infatti devono fare delle terapie chelanti per rimuovere i metalli in eccesso, comunque ci può essere un sovraccarico di ferro che porta a emocromatosi o emosiderosi negli organi parenchimali soprattutto nel cuore e nel fegato.
La secrezione di EPO nel quadro di un’anemia grave è scompensata e porta ad una massiva iperplasia eritroide nel midollo osseo e a un’estesa emopoiesi extramidollare. La massa in espansione dei precursori dei globuli rossi, erode la corticale ossea compromettendo l’accrescimento osseo e causando anomalie scheletriche. L’emopoiesi extramidollare interessa fegato, milza e linfonodi, e in casi estremi produce masse extra ossee nel torace in addome e pelvi.
Possiamo distinguere 3 tipi di β-talassemie:

- Talassemia maior (maggiore) o morbo di Cooley o anemia mediterranea: omozigosi β+/β+ o β0/β0 che richiede trasfusione. È la più grave, soprattutto l’omozigosi β0/β0;
- Talassemia minor (minore) o anemia microcitica familiare o tratto talassemico: eterozigosi β+/β o β0/β. In questo caso il 50% delle catene funzionanti riesce a compensare e la patologia non si manifesta, infatti sono soggetti sani che, come in caso di anemia falciforme, mostrano resistenza al Plasmodium della malaria.
- Talassemia intermedia: eterozigosi β+/β0 o anche varianti lievi di β+/β+, caratterizzata da grave anemia che però non necessita di trasfusioni.
Paradossalmente un deficit nel gene α diminuisce la gravità delle β-talassemie proprio perché precipita un numero minore di catene α e quindi la vita media dei globuli rossi è più elevata.
Caricamento….